Trastornos de las neuronas motoras: causas, síntomas, factores de riesgo, diagnósticos y tratamientos
- Inicio
- Comité Editorial
- Lineamientos
- Carta de Cesión de Derechos
- Información Legal
- Acerca de la Revista
- Bases de Datos
- Contacto
- ISSN 2007-3054
- Centro de Investigaciones Cerebrales
Universidad Veracruzana
Laureani Fierro Ángel de Jesus1, Lara Aparicio Sandra Yasbeth1, Morgado-Valle Consuelo2, Beltrán-Parrazal Luis2, García Luis Isauro2, Hernández María Elena2, Manzo Jorge2, Pérez César Antonio2*
1Doctorado en investigaciones cerebrales; 2Instituto de investigaciones cerebrales. Universidad Veracruzana. Xalapa, Ver.
Resumen
Abstract
Introducción
Desarrollo
Metodología: Criterios de selección
Esclerosis lateral amiotrófica
Esclerosis lateral primaria (ELP)
Atrofia muscular espinal (AME) o la enfermedad de Werdnig-Hoffmann
Síndrome de Guillain-Barré (SGB)
Atrofia muscular espinal-bulbar
Síndrome pospoliomielítico (SPP)
Perspectivas
Conclusiones
Conflicto de intereses
Agradecimientos
Referencias
Mail
En el Sistema Nervioso Central existen diferentes tipos de neuronas. Una de ellas son las células motoras o motoneuronas. Estas células son de gran importancia ya que llevan la información a los músculos del cuerpo. Debido a la degeneración de neuronas motoras piramidales en la corteza motora, neuronas del tallo cerebral y neuronas del asta anterior en la médula espinal inducen la aparición de enfermedades motoras que producen atrofia progresiva de los músculos. Así, cuando se dañan estas células nerviosas, se presentan diferentes enfermedades motoras “raras” como: la esclerosis lateral amiotrófica, la esclerosis lateral primaria, la atrofia muscular espinal, síndrome de Guillain-Barré, la atrofia muscular espinal-bulbar y el síndrome pospoliomielítico, entre otros. Las patologías motoras pueden tener un origen congénito, genético o ambiental (viral, por fármacos o esporádico). Aunque estas enfermedades motoras se consideran poco comunes, son de gran importancia ya que los pacientes no logran recuperarse por la progresión de la enfermedad, lo que conlleva al fallecimiento. Esta revisión, analiza una serie de estudios clínicos de los últimos 20 años en estas patologías motoras para comprender mejor su etiología, diagnóstico y tratamientos, como el uso de células madre en la producción de oligodendrocitos que producen mielina, el cual es un factor clave en las enfermedades motoras y la utilización de antioxidantes que pueden ser la clave para obtener resultados prometedores que tiene como objetivo mantener la esperanza de vida en los sujetos que las padecen.
Palabras clave: Neuronas motoras, enfermedades motoras esporádicas, virus, factores genéticos.
In the Central Nervous System, there are different types of neurons. One of them is motor cells or motor neurons. These cells are of great importance as they carry information to the muscles of the body. Due to the degeneration of pyramidal motor neurons in the motor cortex, neurons of the brain stem and neurons of the anterior horn in the spinal cord induce the appearance of motor diseases that cause progressive muscle atrophy. Thus, when these nerve cells are damaged, different “rare” motor diseases occur, such as amyotrophic lateral sclerosis, primary lateral sclerosis, spinal muscular atrophy, Guillain-Barré syndrome, spinal-bulbar muscular atrophy, and post-polio syndrome, among others. Motor pathologies can have a congenital, genetic, or environmental origin (viral, drug-based, or sporadic). Although these motor diseases are considered rare, they are of great importance since patients fail to recover due to the progression of the disease, which leads to death. This review analyzes a series of clinical analyzes of the last 20 years of these motor pathologies to better understand their etiology, diagnosis, and treatments, such as the use of stem cells in the production of oligodendrocytes that produce myelin, which is a key factor in motor diseases and the use of antioxidants that may be the key to obtaining promising results that aim to maintain life expectancy in those who suffer from them.
Keywords: Motoneurons, sporadic motor diseases, viruses, genetic factors.
El sistema nervioso se divide en dos grandes subsistemas: sistema nervioso central (SNC) y sistema periférico (SNP). El SNC contiene diferentes tipos de neuronas. Un tipo de ellas son las neuronas motoras. Estas células son de gran importancia ya que son las que llevan la información motora del sistema nervioso central a los músculos para que se contraigan o se relajen, por lo tanto, son vías eferentes que hacen sinapsis con las fibras musculares tanto esquelética, visceral y cardíaca.
.Además, este sistema nervioso central está organizado por diferentes tejidos como la corteza cerebral, el cerebelo, el tallo cerebral y la médula espinal. Cada estructura tiene como objetivo regular y controlar el funcionamiento de los diversos órganos y sistemas, coordinando su interrelación y la relación del organismo con el medio externo. Particularmente en la médula espinal existen células que reciben, integran y correlacionan distintos tipos de información sensorial, motora y autonómica. Las neuronas motoras se clasifican en dos tipos: las neuronas superiores que transmiten mensajes desde la corteza motora hasta llegar a las neuronas inferiores que se encuentran en el tallo cerebral y médula espinal para controlar la función de los músculos.
Se sabe que existen diferentes patologías de las neuronas motoras que pueden ser hereditarias o adquiridas. Cuando existe una patología todas las señales de las neuronas motoras son interrumpidas, lo cual puede provocar una espasticidad y rigidez de los músculos, hasta llegar a una atrofia muscular en donde se pierden todos los movimientos voluntarios. Esta revisión describe algunas patologías motoras raras principales de las neuronas motoras como son: la esclerosis lateral amiotrófica, la esclerosis lateral primaria, la atrofia muscular espinal, síndrome de Guillain-Barré, la atrofia muscular espinal-bulbar y el síndrome pospoliomielítico, así como su posible etiología y tratamientos, debido a que tomar en cuenta sólo las manifestaciones clínicas pueden conducir a confusiones. Así, es conveniente realizar un buen diagnóstico utilizando diferentes técnicas y herramientas que ayuden a un mejor resultado en los tratamientos de las distintas enfermedades motoras de sujetos que las padecen.
Los trastornos motores producen movimientos involuntarios o anormales, y deterioro en los voluntarios. Algunos de estos padecimientos neurológicos muy conocidos son la enfermedad de Parkinson, la ataxia espinocerebelosa, la enfermedad de Huntington, el síndrome de Tourette, etc. Se sabe, que los trastornos motores pueden tener como consecuencia, trastornos cognitivos y psiquiátricos con una amplia gama de síntomas. En la mayoría de estos trastornos motores, se pueden tratar los síntomas para mejorar la calidad de vida de las personas que los padecen, ya que hasta la fecha no tienen cura.
Existen otras enfermedades motoras que se les considera como “raras” porque son de baja prevalencia. Esto ha propiciado el poco interés para la política sanitaria y para la investigación. Sin embargo, se debe considerar su impacto en la sociedad porque personas con enfermedades motoras presentan también un deterioro cognitivo, tal es el caso de las personas que padecen esclerosis lateral amiotrófica. Entre las afectaciones cognitivas, las áreas más frecuentemente son la velocidad de procesamiento de información, memoria de trabajo, memoria visual, verbal, lenguaje (fluencia verbal) y funciones ejecutivas. La prevalencia del deterioro cognitivo de pacientes con esclerosis múltiple fluctúa entre el 40 al 70%. Esta disfunción cognitiva puede interferir con las actividades diarias, que implican la interacción social, las relaciones familiares y la capacidad para realizar tareas domésticas; y es la responsable de que los pacientes tengan dificultad para conseguir y mantener un empleo.1
En la mayoría de las enfermedades motoras (en donde están afectadas principalmente las neuronas motoras) son raras por lo que hay una prevalencia por debajo de los 50 en cada 100,000 habitantes por año.2 En México hay poca prevalencia de estas enfermedades motoras. Sin embargo, se siguen presentando en la actualidad (gráfica 1).3-8 Todas estas patologías conllevan a una incapacidad tanto para los adultos como para los niños, en cuanto a su margen de edad es amplio, por lo que en un adulto puede aparecer desde la cuarta década de edad y en un niño puede iniciar desde su nacimiento o antes de que comience a caminar.
Así, las enfermedades de las neuronas motoras representan un grupo heterogéneo de trastornos neurodegenerativos letales cuyas causas aún se desconocen en gran medida. Se calcula que en México hay 6,000 casos de esclerosis lateral amiotrófica (ELA) diagnosticados.9 Las enfermedades motoras producen atrofia progresiva de los músculos debido a la degeneración de neuronas piramidales en la corteza motora, neuronas del tallo cerebral y neuronas del asta anterior en la médula espinal.10
En las enfermedades motoras raras se caracterizan por un deterioro progresivo de las neuronas que inician el movimiento muscular. Como resultado de este deterioro, los músculos se debilitan y ya no pueden funcionar con normalidad. De esta manera, una de las enfermedades motoras es la esclerosis lateral amiotrófica (ELA), la cual es una enfermedad degenerativa de las neuronas motoras principalmente, afectando la neurona motora superior en el homúnculo motor de la corteza cerebral y a la neurona inferior en la medula espinal.11,12 Algo semejante ocurre en la esclerosis lateral primaria (ELP) que se caracteriza a diferencia de la ELA, por un inicio insidioso de disfunción progresiva de la neurona motora superior en ausencia de signos clínicos de afectación de la neurona motora inferior.13 La atrofia muscular espinal (AME) es una enfermedad monogénica por la cual más del 95% de los casos se deben a deleciones o mutaciones dentro del gen de supervivencia de la neurona motora 1 (SMN1).14 La atrofia muscular espino-bulbar es una patología de la neurona motora inferior, ligada al cromosoma X la cual evoluciona lenta y progresivamente.15 El síndrome de Guillain-Barré se manifiesta de manera clínica como una parálisis arrefléxica aguda en donde su evolución puede llegar a los músculos de la respiración y provocar la muerte en los pacientes. Hay que mencionar también, el síndrome pospoliomielítico en donde el virus de la polio ataca específicamente a las neuronas motoras del asta anterior de la médula espinal y del tallo cerebral.
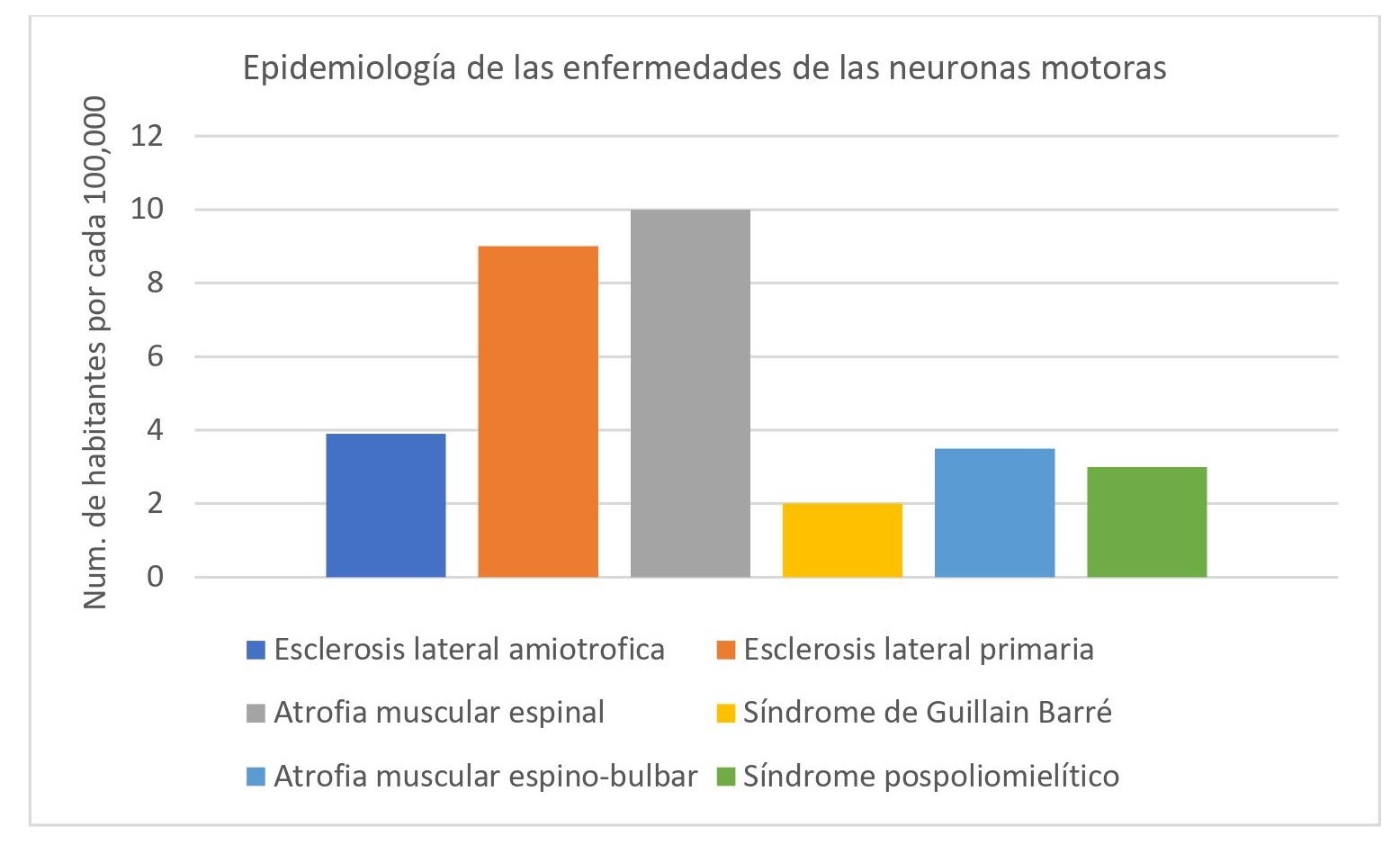
Grafica 1. Prevalencia de las enfermedades motoras raras en México.
3. Metodología: Criterios de selección
En la presente revisión se analizaron artículos científicos que estuvieran relacionados con enfermedades de las neuronas motoras. Se siguieron las recomendaciones PRISMA-P para realizar la revisión sistemática. Para ello, se realizaron búsquedas en las bases de datos electrónicas PubMed, Google Scholar, y SciELO. En la búsqueda se identificaron estudios en personas y animales. Se utilizaron las siguientes palabras clave en idioma español como en inglés: enfermedades de las neuronas motoras, “motor neuron diseases”, esclerosis lateral amiotrófica, “Amyotrophic Lateral Sclerosis”, esclerosis lateral primaria, “primary lateral sclerosis”, atrofia muscular espinal, “spinal muscular atrophy”, enfermedad de Werdnig-Hoffmann, “Werdnig-Hoffmann disease”, síndrome de Guillain-Barré, “Guillain Barre syndrome”, atrofia muscular espino-bulbar, “spino-bulbar muscular atrophy”, síndrome pospoliomielítico, “post-polio syndrome". Se registraron 2025 artículos del año 2001 a septiembre de 2021.
De estos artículos, los criterios de selección para la presente revisión fueron los siguientes:
Población de estudio: Se determinaron de acuerdo con el número total de los artículos que contenían las palabras clave. Criterios de inclusión: artículos que se relacionaron y contaron con las palabras clave de la presente revisión. Criterios de exclusión: artículos que no se relacionaban con el tema de investigación, que el título tampoco estuviera relacionado y artículos que estuvieran duplicados. Muestra: Se determinaron de acuerdo con los artículos que hayan cumplido con los criterios de inclusión (n= “x” número de artículos). Se procedió a la revisión de los artículos incluidos, luego se procedió a la descripción y análisis de éstos para obtener las conclusiones de la presente revisión.

Figura 1. Diagrama de flujo del procedimiento para identificar los artículos que cumplieron los criterios de elegibilidad y que fueron incluidos en esta revisión.
4. Esclerosis lateral amiotrófica
La esclerosis lateral amiotrófica (ELA) es una patología que se caracteriza por la degeneración progresiva de las neuronas motoras superior (NMS) e inferior (NMI), lo que provoca debilidad de los músculos de las extremidades, torácicos, abdominales y bulbares.16 La ELA tiene dos divisiones; ELA familiar y ELA esporádica, en donde la ELA familiar se presenta entre un 5 a 10% de los casos mientras que la ELA esporádica se presenta en un 90 a 95%. Antes de los 65 años, la esclerosis lateral amiotrófica es más común en hombres que en mujeres.17
4.1. Causas
Se han propuesto diferentes teorías del origen de la ELA. Una de ellas hace referencia a las mitocondrias que son un orgánulo importante de la célula pues es donde se lleva a cabo la “respiración” y la generación de ATP. Se ha investigado y se ha visto que la dinámica mitocondrial, el transporte axonal y el control de calidad mitocondrial están entrelazados en un papel fundamental en la homeostasis de la red mitocondrial neuronal, en la salud y la enfermedad de la genética de la ELA familiar y sugieren que estos procesos se ven fuertemente afectados en todo el curso de la enfermedad, pero que aún se necesitan hacer más investigaciones para lograr una mejor compresión detallada.18
Por otro lado, el estrés oxidativo (OS) es una condición producida por el desequilibrio entre oxidantes y antioxidantes en un sistema biológico. El desequilibrio se produce como resultado del nivel excesivo de especies reactivas de oxígeno (ROS por sus siglas en inglés) o del funcionamiento inadecuado del sistema antioxidante, existe un control homeostático en la célula para verificar el nivel de ROS en condiciones normales, la ELA familiar en un bajo porcentaje de los casos son el resultado de una mutación en el gen responsable de codificar la superóxido dismutasa, mientras que la ELA esporádica en un alto porcentaje no tiene conexión genética clara, la mutación genética más común es identificado como la repetición expandida de hexanucleótidos GGGGCC en la región no codificante del gen C9ORF72 en el cromosoma 9p21, hay informes de varios factores en la ELA, como pueden ser la excitotoxicidad, disfunción mitocondrial, estrés del retículo endoplásmico y neuroinflamacion.19 Otro metabolito antioxidante, es el ácido úrico, donde se demostró que puede estar implicado en la fisiopatología de la ELA, ya que en aumento de los niveles de ácido úrico se asocia con la progresión de la enfermedad.20
Otra de las causas que se le podrían atribuir es la autofagia en la microglía, en un estudio donde se analizó la disfunción de la autofagia especifica de la glía en ELA, se observó que la pérdida de la función de C9orf72 enfatizó el papel de la autofagia de la microglía en la ELA, la deleción de C9orf72 en ratones no dio lugar a un fenotipo de la neurona motora, si no que dio lugar a una acumulación lisosomal y una respuesta inmune alterada en la microglía, pero aún no está claro si la insuficiencia de C9rof72 es una causa primaria de ELA, aunque la sobreexpresión de una forma mutada de superóxido dismutasa 1 (SOD1) en microglía dio una idea más detallada del papel de la autofagia en la estas células durante la ELA.21
Las investigaciones sobre la ELA han ido en aumento con el paso del tiempo, pero no se ha descrito exactamente la etiología de esta patología, ya que como se mencionó anteriormente casi el 95% de los casos son esporádicos en donde no se ha logrado homogenizar en una teoría, por lo que en su mayoría es en la ELA familiar donde se han identificado diferentes tipos de genes en las últimas décadas.
4.2. Síntomas
La ELA presenta distintos síntomas a lo largo de la enfermedad, uno de los principales es la debilidad muscular progresiva, como también atrofia muscular, fasciculaciones, calambres musculares,22 espasticidad muscular con dificultad para hablar (disartria), disfagia e incluso dificultad respiratoria, la cual puede llegar a causar la muerte.23
4.3. Factores de riesgo
Se han investigado diferentes factores de riesgo para el desarrollo de la ELA, se han propuesto diferentes teorías en las cuales se ha propuesto el tabaquismo ya que el humo del cigarrillo aumenta el riesgo por diferentes mecanismos que incluyen inflamación, estrés oxidativo y neurotoxicidad a causa de los metales pesados y compuestos químicos que surgen en el humo.24
4.4. Diagnóstico
Se han utilizado diferentes técnicas para el diagnóstico de la ELA, por lo que se debe conocer completamente la enfermedad. Esto ha llevado años llegar al punto en que nos encontramos ahora. Una de esas técnicas que se utilizan para realizar el diagnostico son los biomarcadores moleculares para los diferentes procesos patológicos como pueden ser la excitotoxicidad, el estrés oxidativo, la disfunción mitocondrial y la neuroinflamación.23
Los estudios de Electrodiagnóstico son la clave para establecer la certeza diagnóstica y facilitar el diagnóstico precoz en la ELA al considerar la evidencia electrofisiológica como un equivalente de los signos y síntomas clínicos. La cuantificación y puntuación de las anormalidades electromiográficas puede ser útil como medida complementaria en la descripción y clasificación (nivel de certeza diagnóstica) de la alteración de la neurona motora inferior en los pacientes con ELA. Los criterios electromiográficos utilizados actualmente permiten clasificar en niveles de certeza diagnóstica de acuerdo con hallazgos compatibles con denervación activa o reinervación crónica; sin embargo, se presentan casos en los que no se logra cumplir con la totalidad de los criterios y para que se cumplan se requiere extender el examen (mayor cantidad de músculos examinados) lo que puede dificultar su uso durante la práctica clínica.25
La disartria y la atrofia de lengua con fasciculaciones son datos clínicos prominentes que se presentan en ELA, las anormalidades en la lengua se presentan por daño en el nervio hipogloso en bulbo raquídeo produciendo disartria flácida que caracteriza a la disfunción de la NMI. Estudios previos han descrito en vista sagital de la imagen por resonancia magnética (IRM) de encéfalo un hallazgo incidental denominado “signo de lengua brillante” en pacientes con ELA. Este signo se ha asociado al proceso degenerativo de la lengua y se ha propuesto como un signo radiológico útil en la evaluación diagnóstica de estos pacientes.26
En 1990 la Federación Mundial de Neurología estableció los criterios diagnósticos conocidos como criterios de El Escorial (Los criterios diagnósticos de El Escorial se basan en la definición de regiones anatómicas: bulbar, cervical, torácica y lumbosacra). De acuerdo con estos criterios el diagnostico de ELA requiere signos de neurona motora superior, así como de signos de neurona motora inferior, y un curso progresivo. Además de estos criterios clínicos se debe realizar varias pruebas tales como: una prueba de velocidad de conducción de nervios, una biopsia del músculo para detectar si existe una atrofia de las fibras musculares. Asimismo, el médico también puede solicitar un análisis de sangre y de orina para descartar la posibilidad de otras enfermedades neurológicas.27
4.5. Tratamientos
En la actualidad, solo hay 2 fármacos que se encuentran disponibles para el tratamiento paliativo de la ELA, riluzol y edaravona. Se ha propuesto que este medicamento actúa inhibiendo los procesos de glutamato; este medicamento es rápidamente absorbido después de su administración oral y se distribuye ampliamente en todo el cuerpo. Se ha demostrado que cruza la barrera hematoencefálica. En cuanto a su vía de administración: la dosis diaria recomendada para adultos y pacientes de edad avanzada es 100 mg al día.28 El otro fármaco para el tratamiento de la ELA es el edaravona (Radicava), el cual es un potente antioxidante que evita que el estrés oxidativo induzca la muerte de las neuronas motoras, también es un eliminador de radicales libres que inhibe la nitración de los residuos de tirosina en líquido cefalorraquídeo, se administra 60mg mediante infusión intravenosa muy lenta aprox. 60 minutos en ciclos de 28 días.29 El mecanismo de acción del primero de ellos, el riluzol, se basa en el bloqueo de los canales controlados por voltaje en las neuronas presináptica, lo que se traduce en una reducción de transmisión glutamatérgica. En los ensayos realizados, se ha observado que, aunque aumenta la supervivencia hasta los 3 meses no es capaz de ejercer ningún efecto sobre la fuerza del músculo. El medicamento edavarona, el cual está aprobado únicamente por la FDA (Administración de Alimentos y Medicamentos de los Estados Unidos), que actúa como agente antioxidante, es capaz de retrasar la progresión de la enfermedad en pacientes seleccionados de forma restringida. Estos dos fármacos (riluzol y edavarona), no son capaces de proporcionar un tratamiento efectivo, por lo que se hace necesario e indispensable el descubrimiento y desarrollo de nuevos fármacos que frenen el avance de la enfermedad.30
Por otra parte, es importante realizar un adecuado abordaje el cual no solo es el tratamiento farmacológico, como pueden ser la terapia física: asociada a equipamientos especiales, estos en conjunto, pueden aumentar la independencia durante el curso de la ELA. El ejercicio aeróbico suave y de bajo impacto, como caminar, nadar y montar bicicleta fija, pueden fortalecer los músculos no afectados, mejorar la salud cardiovascular, así como combatir la fatiga y la depresión. A su vez, hay terapia de lenguaje ya que los pacientes con ELA que desarrollan problemas de dicción que se le conoce como disartria. Este trastorno puede beneficiarse de trabajar con un terapeuta del lenguaje, quien les puede enseñar estrategias para hablar más fuerte y más claro, y de esta manera mantener la habilidad para comunicarse. El siguiente punto es de soporte nutricional ya que es esencial, pues se ha demostrado que estos pacientes se debilitan y pierden peso, la educación nutricional dirigida hacia el paciente y sus familiares, facilita la alimentación para que se les pueda proporcionar suficientes calorías, fibra y líquidos, así como evitar alimentos que sean difíciles de digerir. Cuando ya no se puede obtener suficiente nutrición a través de la vía oral, se debe valorar la necesidad de una gastrostomía endoscópica percutánea.31,32
5. Esclerosis lateral primaria (ELP)
La esclerosis lateral primaria es un trastorno de la neurona motora superior progresivo, que produce espasticidad espino-bulbar, con una diferencia con la ELA en que este trastorno no daña a las neuronas motoras inferiores.33 Este trastorno es muy raro y solo representa aproximadamente del 1 al 4% de todos los pacientes con alguna enfermedad de la neurona motora.13 Esta afección generalmente ocurre entre los 40 y los 60 años, y es más frecuente en hombres que en mujeres.17
5.1. Causas
La ELP se caracteriza por el mal funcionamiento de los tractos corticoespinales descendentes, por lo cual es la predominante degeneración de células piramidales. Como lo muestran en un estudio longitudinal de neuroimagen de la ELP en donde hubo una reducción total del volumen del tronco encefálico que se debe principalmente a la patología del bulbo raquídeo, pero los cambios pontinos también contribuyen a la atrofia del tronco encefálico, por lo que la ELP presenta una atrofia medular, pontina y mesencefálica en comparación con la ELA, por lo que estas reducciones de volumen son causadas principalmente por la degeneración de la neurona motora superior.34 Fisiológicamente, aún no se ha comprendido exactamente las causas, no obstante, hay una teoría que es secundario a mutaciones en el gen ALS2, lo que lleva a la inestabilidad de las proteínas y la ausencia de una función normal.35
La esclerosis lateral primaria se divide en dos: la esclerosis lateral primaria juvenil la cual se relaciona con las mutaciones del gen ALS2 y la esclerosis lateral primaria en adultos en donde se desconoce la causa evidente. En la mayoría de los casos, no es una enfermedad hereditaria y no se sabe por qué o cómo comienza.17
5.2. Síntomas
Los síntomas de la esclerosis lateral primaria, se pueden confundir con la esclerosis lateral amiotrófica, la diferencia es que, en la esclerosis lateral primaria, por lo general, tardan años en progresar. Por ejemplo, hay rigidez, debilidad y espasmos musculares (espasticidad) en las piernas, que a menudo comienza en una sola pierna, conforme avance la enfermedad afectará el tronco, posteriormente los brazos, las manos, la lengua y la mandíbula, lo que ocasionará disfagia, también se presentará bradicinesia, ronquera, afasia y babeo a medida que los músculos faciales se debilitan y en las últimas etapas de la enfermedad habrá dificultades para respirar.17
5.3. Factores de riesgo
La esclerosis lateral primaria es una enfermedad la cual no tiene una etiología clara, por lo que no hay factores de riesgo que se hayan documentado como posibles, no posee relación familiar, aparece aleatoriamente principalmente en la quinta década de vida y sin razón alguna.
5.4. Diagnóstico
El diagnóstico para la esclerosis lateral primaria es por exclusión de patologías que causan un padecimiento muy semejante como lesiones compresivas de la médula espinal, sífilis, enfermedad de Lyme, esclerosis múltiple, forma mielopática de la adrenoleucodistrofia, entre otras.36
Después de registrar el historial médico y familiar, las pruebas para detectar la ELP (y así descartar otras afecciones) incluyen:
Resonancia magnética. Una resonancia magnética u otras pruebas de diagnóstico por imágenes del cerebro o la columna vertebral puede revelar signos de degeneración de las células nerviosas, una resonancia magnética puede mostrar otras causas de los síntomas, como anomalías estructurales, esclerosis múltiple o tumores de la médula espinal.
Electromiograma. Durante un electromiograma, un médico inserta un electrodo de aguja a través de la piel en varios músculos. El análisis evalúa la actividad eléctrica de los músculos cuando se contraen y cuando están en reposo. Esta prueba puede medir el compromiso de las neuronas motoras inferiores, lo que puede ayudar a diferencias entre esclerosis lateral primaria y esclerosis lateral amiotrófica.
Estudios de la conducción nerviosa. Estas pruebas utilizan una cantidad baja de corriente eléctrica para medir la capacidad de los nervios para enviar impulsos a los músculos en distintas áreas del cuerpo. Esta prueba puede determinar si tienes daño en los nervios.
Punción lumbar. Un médico utiliza una aguja delgada y hueca para extraer del canal raquídeo pequeñas muestras del líquido que rodea el cerebro y la médula espinal (líquido cefalorraquídeo) para realizar análisis de laboratorio. Una punción lumbar puede ayudar a descartar esclerosis múltiple, infecciones y otras afecciones.17
5.5. Tratamientos
En el tratamiento de la esclerosis lateral primaria, solo se utilizan medicamentos que puedan reducir la espasticidad y antidepresivos tricíclicos para la afección pseudobulbar.36
Se pueden recetar medicamentos como baclofeno (40-80 mg/día), tizanidina (2 mg/día) o clonazepam (<20 mg/día) para aliviar los espasmos musculares (espasticidad). Estos medicamentos se administran por vía oral. Si la espasticidad no se controla con medicamentos orales, se puede realizar una implantación quirúrgica de una bomba de medicamentos para administrar baclofeno directamente al líquido cefalorraquídeo (baclofeno intratecal), la amitriptilina también puede ayudar con problemas de babeo.37
Por otra parte, existen otros tratamientos como la fisioterapia en los cuales hay ejercicios de estiramiento y fortalecimiento que pueden ayudar a mantener la fuerza muscular, la flexibilidad y la amplitud de movimiento, y a prevenir la inmovilidad de las articulaciones. La terapia del habla ayuda ya que los músculos faciales se pueden ver afectados.17
6. Atrofia muscular espinal (AME) o la enfermedad de Werdnig-Hoffmann
La atrofia muscular espinal se refiere a un grupo de trastornos genéticos, todos caracterizados por la degeneración de las neuronas del asta anterior de la médula espinal, la atrofia y la debilidad muscular resultante. Es un trastorno autosómico recesivo que resulta de una deleción o mutación homocigótica en el gen de supervivencia de la neurona motora (SMN1, en inglés Survival Motor Neuron 1).38 Se estima una prevalencia de 1/70.000 aproximadamente. La enfermedad es ligeramente más frecuente en hombres que en mujeres.39
6.1. Causas
La causa más frecuente es debida a la alteración del gen de supervivencia de la neurona motora (SMN1), localizado en la región cromosómatica 5q13, que codifica para la proteína SMN. El 95 a 98% de los pacientes presentan ausencia en homocigosis del gen SMN1, detectable por estudio molecular del exón 7 y que confirma genéticamente el diagnostico de AME.40 Otros tipos menos comunes de la AME pueden deberse a cambios en otros genes.
6.2. Síntomas
La AME tiene síntomas variados y por ello, se clasifica en 4 diferentes tipos de acuerdo con la gravedad de la enfermedad. Tipo I (se subdivide en Ia y Ib) es el tipo más común y el más grave de la AME, el curso de la enfermedad implica deterioro de la primera infancia ya que sus inicios van desde el periodo prenatal hasta los 6 meses de vida, los pacientes con AME tipo I nunca adquieren la capacidad de sentarse sin apoyo. La AME tipo II se caracteriza por el inicio a una temprana edad ya que desde los 18 meses puede aparecer y tiene un mayor rango de supervivencia que el tipo I. Los pacientes con AME II logran la capacidad de estar sentados y, rara vez, de pie, pero no logran caminar. La AME tipo III es una forma más leve con inicio desde el primer año hasta la tercera década de vida, en este tipo es característico una progresión lenta, la capacidad de estar de pie y caminar a menudo se conservan hasta la edad adulta, este tipo de AME se subdivide en (IIIa y IIIb). Y por último la AME tipo IV que es la adulta tiene un inicio tardío dentro de la segunda y tercera década y tiene una esperanza de vida normal, los pacientes muestran solo un caminar afectado.41
Otros síntomas que se presentan incluyen contractura persistente de las articulaciones con postura anormal fija del miembro, se logran manifestar contracturas graves, escoliosis, deformidad del tórax, problemas respiratorios, mandíbulas inusualmente pequeñas, caída de los parpados superiores, además se puede presentar debilidad y atrofia de los músculos de la cara, la mandíbula y la lengua, llevando problemas al masticar, deglutir y cambios en el habla, dolor muscular y fatiga. Puede haber pérdida sensorial en las manos y los pies, y agrandamiento de las mamas masculinas.42
6.3. Factores de riesgo
La atrofia muscular espinal es la enfermedad neuromuscular hereditaria con mayor índice de mortalidad en la infancia, la incidencia varía entre 1 por cada 6000 a 11000 casos en nacidos vivos, se desconocen otros factores de riesgo.
6.4. Diagnóstico
Cuando se sospecha que un paciente puede tener AME un análisis genético puede confirmar el diagnóstico, como también la identificación de familiares que han sido afectados y la detección de portadores de la enfermedad, entre otros estudios.
El análisis del ADN por amplificación selectiva (PCR) de los exones 7 y 8 del gen SMN1 mostrará la ausencia (deleción homocigota) del gen SMN2. Dicho resultado confirma el diagnostico de AME por afectación del gen SMN1 en un 95% de los casos. Cuando el análisis genético no ayuda a la confirmación del diagnóstico, se puede recurrir a una biopsia muscular, la cual muestra un patrón neuropático característico, con fibras hipertrofiadas que normalmente muestran propiedades histoquímicas de fibras tipo 1 (lentas) y fibras pequeñas de forma redondeada.43
6.5. Tratamientos
Hasta la fecha actual no existe una cura para la AME. Sin embargo, la comprensión de la genética molecular de la AME ha llevado al desarrollo de modelos preclínicos y numerosos enfoques terapéuticos potenciales.38,44-47 Diversas investigaciones han demostrado que la calidad de vida puede mejorar significativamente y la progresión de la enfermedad se puede ralentizar con una intervención oportuna. Los pacientes con AME requieren asistencia médica de por vida para el manejo adecuado de problemas ortopédicos, respiratorios y nutricionales.48 Los familiares de los pacientes también necesitan educación para mejorar la calidad de vida del paciente.
Existen medicamentos recientes como el nusinersen (Spinraza) (dosis 12mg, vía punción lumbar) aprobado por la FDA en diciembre de 2016 para tratar a niños y adultos con atrofia muscular espinal. nusinersen es una inyección administrada en el espacio donde está el líquido que rodea la médula espinal. Se usa para los tipos I, II, III y IV de la AME. Los estudios han mostrado que el tratamiento continuo con nusinersen aumenta la función motora y ralentiza la progresión de los síntomas. En general, el uso del medicamento disminuye los problemas respiratorios, de nutrición y la necesidad de ingresos hospitalarios. Sin embargo, la respuesta al tratamiento varía y algunas personas pueden no responder al medicamento en absoluto o pueden tener complicaciones médicas que impidan el uso del tratamiento.42
El risdiplam es otro medicamento aprobado por la FDA en agosto de 2020 fue aprobado para el tratamiento de la AME en pacientes de 2 meses de edad o más. Este fármaco es un modificador de empalme de pre-ARNm de SMN2 de molécula pequeña, (dosis diaria 0.20-5 mg/kg, vía oral).42,49
Así mismo, hay otro medicamento que es el onasemnogene abeparvovec (volumen total de dosis 16.5-115.5 ml, vía IV), anteriormente conocido como AVXS-101 (y comercializado con el nombre de Zolgensma) fue aprodabo por la FDA en mayo del 2019, para el tratamiento de pacientes pediátricos menores de 2 años con AME con mutaciones bialélicas en el gen de la neurona motora de supervivencia 1 (SMN1). El Zolgensma es una suspensión de una terapia génica basada en vectores virales adenoasociados para infusion intravenosa. Es un AAV9 autocomplementario recombinante que contiene un transgén que codifica la proteína de la neurona motora de supervivencia, bajo el control de un potenciador de citomegalovirus/ promotor híbrido de β-actina de pollo.50
7. Síndrome de Guillain-Barré (SGB)
El síndrome de Guillain Barré, está caracterizado por una debilidad muscular progresiva, cuya expresión clínica más conocida es la parálisis arrefléxica aguda y una disociación albúmino-citológica en el líquido cefalorraquídeo.51 Dicha debilidad está mediada por un mecanismo autoinmune a nivel del nervio periférico, la magnitud de la debilidad es variada, desde leve hasta completa cuadriplejia flácida con compromiso de los músculos de la respiración hasta en un 30% de los casos.52 El síndrome de Guillain-Barré puede afectar a personas de todas las edades. Sin embargo, el riesgo aumenta a medida que se envejece. También es más común en hombres que en mujeres.53
7.1. Causas
El SGB es una polineuropatía inflamatoria aguda de rápida evolución, es una enfermedad autoinmune desencadenada por la infección viral o bacteriana que destruye la vaina de mielina de los nervios periféricos.54,55 Algunas infecciones previas virales o bacterianas se han ligado con esta patología; principalmente las de tipo gastrointestinal y respiratorio, como se ha evidenciado en algunos casos. De esta manera, alrededor del 70% de pacientes han reportado un antecedente infeccioso entre la primera y la sexta semana antes del cuadro clínico.56
El aumento en el número de casos de SGB durante el brote de virus de Zika (ZIKV) en el continente americano proporciona evidencia epidemiológica del vínculo entre la infección por ZIKV y el SGB, entre 2015 y 2016, surgieron grupos de SGB poco después del brote del virus de ZIKA (ZIKV) en el continente americano, en un patrón que destacó una asociación temporal y geográfica entre los casos de SGB y la transmisión del ZIKV. Clínicamente predominó entre las regiones afectadas por la epidemia, pero el espectro de enfermedades neurológicas en los adultos parece más amplio, ya que también se ha informado casos de encefalopatía, encefalitis, meningitis, mielitis y convulsiones. Se ha descrito un perfil temporal para-infeccioso de SGB asociado a ZIKV en estudios clínicos, lo que sugiere un efecto neuropático viral directo. Se consideran cambios genéticos virales adaptativos e interacciones inmunológicas con otros flavivirus circulantes y otros factores.57
Entre otros antecedentes patológicos personales se resalta conocer los procedimientos quirúrgicos previos, el lupus eritematosos sistémico y los linfomas que incrementan el riesgo.56
7.2. Síntomas
Las complicaciones agudas de mayor relevancia se encuentran en la afectación de los músculos respiratorios llegando a requerir ventilación mecánica en 25% de los casos. De igual manera se presentan alteraciones autonómicas en un 15% de los pacientes (taquicardia sinusal, hipertensión, arritmias cardiacas e hipotensión postural). También se han mostrado secuelas neurológicas en un 89% de los casos, en especial los síntomas sensitivos.52
7.3. Factores de riesgo
El SGB tiene una amplia distribución mundial, afecta a todas las razas, edades y nacionalidades, se ha observado un aumento de la frecuencia en pacientes mayores de 75 años. La incidencia mundial se encuentra entre 0.6 a 4 casos por cada 100,000 habitantes al año.52
7.4. Diagnóstico
El diagnóstico de esta enfermedad es clínico, basándonos en criterios clínicos mismos que incluyen obligatoriamente: una debilidad progresiva en uno o más miembros debido a una neuropatía, arreflexia, y un curso de la enfermedad menor de 4 semanas y exclusión de otras causas. Otros criterios clínicos incluyen: debilidad simétrica relativa, leve afectación sensorial, alteración de cualquier par craneal, ausencia de fiebre y evidencia electrofisiológica de desmielinización. Existen pruebas complementarias disponibles como el estudio de líquido cefalorraquídeo donde pueden encontrarse proteínas elevadas después de una semana y menos de 10 linfocitos/mm3. Además, en las pruebas electrofisiológicas pueden encontrarse una conducción nerviosa lenta, latencias distales prolongadas y respuestas tardías anormales.56
7.5. Tratamientos
Esta patología constituye una emergencia médica cuyo tratamiento actual se basa en el soporte vital avanzado en la Unidad de Cuidados Intensivos, así como, la administración de gammaglobulina intravenosa y/o sesiones de plasmaféresis, que a pesar de no disminuir la mortalidad, permiten una recuperación de corta duración.56
En la plasmaféresis, la porción líquida de parte de la sangre (plasma) se extrae y se separa de las células sanguíneas. Luego las células sanguíneas se vuelven a colocar en el cuerpo, el cual produce más plasma para compensar lo que se extrajo. La plasmaféresis puede funcionar liberando al plasma de ciertos anticuerpos que contribuyen al ataque del sistema inmunitario a los nervios periféricos.
Para la terapia de inmunoglobulina, esta contiene anticuerpos sanos de donantes de sangre. Se administra a través de una vena (por vía endovenosa). Las dosis altas de inmunoglobulinas pueden bloquear los anticuerpos perjudiciales que podrían contribuir al síndrome de Guillain-Barré.53
8. Atrofia muscular espinal-bulbar
La atrofia muscular espino-bulbar (AMEB), es conocida también como la enfermedad de Kennedy, es una patología de la neurona motora inferior comenzando a nivel espinal y luego en la evolución a nivel bulbar. Es una afectación que está ligada al cromosoma X de la edad adulta caracterizada por una debilidad lenta y progresiva de los músculos bulbar y de las extremidades.15 La AMEB es un trastorno neurodegenerativo raro de herencia que afecta comúnmente a los hombres en la edad adulta.58
8.1. Causas
La patología es causada por una repetición de expansión de trinucleótidos (CAG) en el gen del receptor de andrógenos (AR) en el cromosoma X, por lo que esta mutación da como resultado un tracto de poliglutamina explandido y una ganancia de función tóxico dependientes de andrógenos en la proteína mutante.59
8.2. Síntomas
Los síntomas clínicos más comunes son debilidad, temblor y calambres, a menudo en una distribución asimétrica y bulbar, con atrofia muscular asociada. La aspiración y la disartria también son comunes debido a la afectación de la musculatura bulbar.59
8.3. Factores de riesgo
Esta patología aparece aproximadamente entre los 18 a 64 años de edad, con una preferencia al sexo masculino en donde aparece en la cuarta década de vida.60 Las mujeres son normalmente asintomáticas o pueden presentar calambres musculares periódicos.61 A diferencia de otras patologías como la ELA, el avance de esta patología es lento, con una disminución de la fuerza muscular del 2% por año.60 La gran mayoría de los pacientes tienen una esperanza de vida normal, sin embargo, corren el riesgo de asfixia por alimento debido a la debilidad de los músculos relacionados con la masticación.61
8.4. Diagnóstico
Cuando se sospecha que la enfermedad está presente, se valora la historia clínica del paciente, se le realiza estudios de neuroconducción, electromiografía y análisis molecular (expansión del triplete CAGn).62,63
8.5. Tratamientos
Como se dijo anteriormente, la atrofia muscular espinobulbar es de lenta progresión, por lo que es difícil tener un modelo animal óptimo para el estudio de diferentes tratamientos en busca de la cura. Sin embargo, hay investigaciones en ratones transgénicos en donde muestran los signos de la enfermedad muy rápido, por lo que hay una degeneración y una muerte aproximadamente en 1 o 2 meses desde su inicio, contrarios a otros estudios en donde la enfermedad inicia más tarde, pero tienen una muerte prematura. Por lo que estos ensayos no son óptimos para un excelente tratamiento.64 Actualmente no existe un tratamiento específico para la AMEB. Este es sintomático y consiste en fisioterapia y rehabilitación. El temblor puede beneficiarse del tratamiento con propranolol (inicialmente 40mg de dos a tres veces al día).62 Se debe prevenir las complicaciones secundarias, entre ellas la neumonía aspirativa debido a debilidad bulbar.62 Los pacientes pueden recibir vitamina E, complejo vitamínico B o fisioterapia muscular como parte del tratamiento. Se han realizado ensayos basados en el déficit androgénico en ratones mediante fármacos como el agonista de GnRH leuprorelina, o el inhibidor de la 5-alfa reductasa dutasteride. Se previene así la translocación nuclear de receptor de andrógeno (RA) aberrante, lo que protege de la acumulación tóxica de la proteína mutante del RA y logra suprimir las manifestaciones de deterioro nueromuscular.58
9. Síndrome pospoliomielítico (SPP)
La poliomielitis paralítica es causada por una infección con el poliovirus.65 Los humanos son los únicos seres pródigos de este virus.66 Este virus ataca específicamente a las neuronas motoras del asta anterior de la médula espinal y el tronco encefálico. Su multiplicación es responsable de una apoptosis de la neurona motora afectada. Por otra parte, las neuronas motoras adyacentes vuelven a inervar las fibras musculares huérfanas que se han denervado por la infección aguda de la poliomielitis creando unidades motoras gigantes.
Las descripciones patológicas han sido pocas y han enfatizado la inflamación persistente o nueva en las meninges, la medula espinal y los músculos de los pacientes afectados. La etiología más probable es la degeneración distal de las unidades motoras anormalmente agrandadas que se forman después de la poliomielitis. Por lo tanto, da como resultado una denervación irregular de las fibras musculares.67 La poliomielitis es más probable que ocurra en niños de 4 a 15 años en climas templados.68
9.1. Causas
El poliovirus tiene un receptor común conocido como receptor de poliovirus (PVR) que pertenece a la superfamilia de inmunoglobulinas CD155, un receptor de superficie celular de poliovirus que se encuentra en el cromosoma 19q13.2.65
9.2. Síntomas
El síntoma más frecuente en pacientes fue el dolor músculo-esquelético, seguido de la fatiga y el cansancio. En pacientes se ha presentado la existencia de sintomatología cognitiva, así como atrofia de los músculos afectados.69,70
9.3. Factores de riesgo
Los factores de riesgo, aunque no están claros, algunos estudios proponen que a mayor edad en la fase aguda de la enfermedad, en el sexo femenino, se presenta una mayor gravedad del cuadro motor inicial, un menor grado de recuperación funcional tras la infección aguda, una mayor duración del periodo de latencia desde el cuadro agudo de la enfermedad hasta el inicio de la recuperación, el uso de ventilación mecánica en la fase aguda de la enfermedad, y un grado importante de actividad física, podrían relacionarse con mayor riesgo de desarrollar un síndrome pospoliomielítico.69
9.4. Diagnósticos
En un estudio en donde se analizaron pacientes con 14 y 16 años después de la primera evaluación con diagnóstico de poliomielitis, en donde el resultado fue que si hay cambios en los síntomas como en la fuerza muscular de los miembros inferiores sobre todo en la flexión y en la marcha. Sin embargo, este cambio es lento en comparación con otros estudios.71
La electromiografía y los estudios de conducción nerviosa. La electromiografía mide las descargas eléctricas diminutas que se producen en los músculos. Se inserta un electrodo de aguja delgada en los músculos que se van a estudiar. Un instrumento registra la actividad eléctrica del músculo en descanso y al contraerlo. En una variación de la electromiografía llamada “estudio de conducción nerviosa”, se adhieren dos electrodos a la piel por encima del nervio a estudiar. Estas pruebas ayudan a identificar y descartar enfermedades como una anomalía de los nervios (neuropatía) y un trastorno del tejido muscular (miopatía).
Otro estudio puede ser por imágenes, mediante exploración por resonancia magnética o por tomografía computarizada para ver imágenes del cerebro y la médula espinal. Estos estudios pueden ayudar a descartar trastornos de la médula espinal, como la espondilosis.
Además, se puede realizar biopsia muscular, que ayuda a buscar evidencia de otra enfermedad que pueda estar causando debilidad y se puede hacer análisis de sangre, las personas con síndrome pospoliomielítico generalmente obtienen resultados de análisis de sangre normales. Los resultados de análisis de sangre anormales pueden indicar otro problema no diagnosticado que este causando los síntomas.17
9.5. Tratamientos
Aun no existe tratamiento farmacológico alguno para el síndrome pospoliomielítico, por lo que el tratamiento que se le da a estos pacientes solo consiste en aminorar los síntomas y dar soporte.70
Una de las técnicas puede ser la conservación de energía, esto implica ajustar tu actividad física y descansar con frecuencia para reducir la fatiga, un terapeuta puede mostrarte formas de respirar que ayudan a conservar la energía. La fisioterapia puede fortalecer los músculos sin fatigarlos. Generalmente consiste en actividades menos extenuantes, como por ejemplo nadar o hacer aeróbicos como los ejercicios acuáticos. También hay terapia del habla, en donde un terapeuta enseña las maneras de compensar las dificultades para tragar, los ejercicios de fortalecimiento de la voz son útiles. Así mismo, hay tratamiento para la apnea del sueño, es posible que se tengan que cambiar los patrones del sueño tales como evitar dormir boca arriba o utilizar un dispositivo que ayude a abrir las vías respiratorias cuando se duerme.17
También se utilizan analgésicos como la aspirina (500mg c/4 o 6 horas, vía oral)17 el paracetamol (600mg-1300mg c/6 horas, vía oral)72 y el ibuprofeno (200mg c/4 o 6 horas, vía oral)73 que pueden aliviar el dolor muscular y articular. Otras posibles opciones de tratamiento pueden incluir el fármaco anticonvulsivo gabapentina (Neurontin, Gralise) (8-35mg/kg/día, vía oral) que se utiliza a menudo para tratar el dolor de los nervios.17
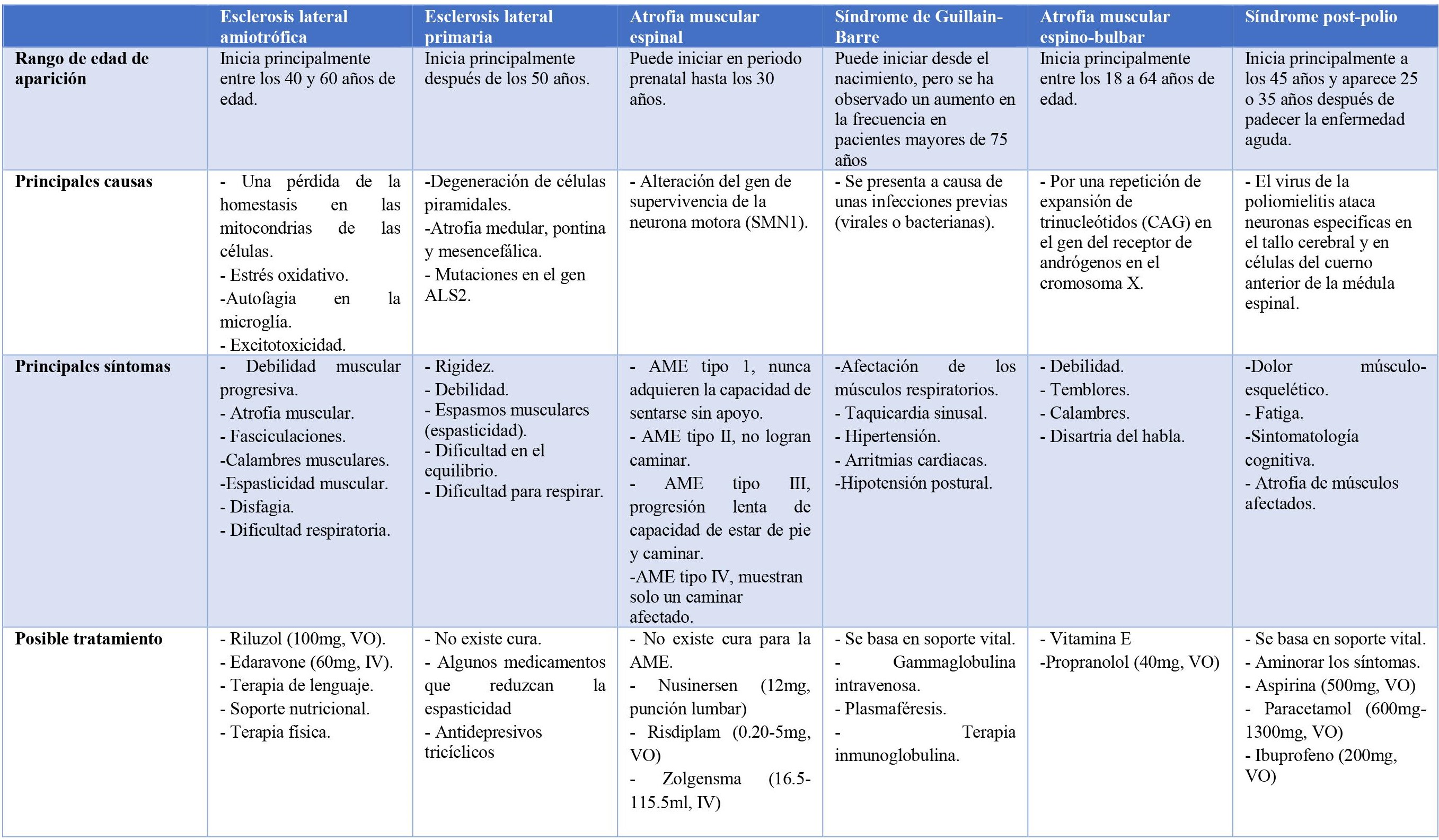
Tabla 1. Tabla comparativa de las enfermedades motoras raras. Información tomada de: 13,18,23,30,32,42,43,50,53,6,62
Las enfermedades motoras evolucionan a un ritmo diferente en cada persona por lo que afectan a los individuos en formas diferentes. En las etapas tempranas de la enfermedad, los síntomas pueden ser similares a los de otras enfermedades, haciendo difícil el diagnóstico. En la actualidad se están realizando investigaciones para comprender las causas de las enfermedades motoras como la esclerosis lateral amiotrófica, la esclerosis lateral primaria, la atrofia muscular espinal, síndrome de Guillain-Barré, la atrofia muscular espinal-bulbar y el síndrome pospoliomielítico.
Algunas personas con estas enfermedades motoras cuentan con antecedentes familiares. Al parecer, esto indica un vínculo hereditario. Aunque generalmente, este tipo de enfermedades aparecen sin motivo en la mayoría de las veces. Esto hace que algunas de las enfermedades motoras que se describieron en el presente artículo se consideren como esporádicas.
No existe una prueba única para diagnosticar a las enfermedades motoras. Normalmente, el diagnóstico se efectúa en base a los síntomas que presentan lo sujetos que la padecen para descartar otras patologías neurológicas. Comúnmente, las pruebas que se utilizan para diagnosticar a las enfermedades motoras incluyen la electromiografía (para medir la actividad eléctrica de los músculos), pruebas de conducción nerviosa (para medir la velocidad en la que se desplazan las señales eléctricas a través de los nervios), la estimulación magnética transcraneal (para medir la actividad de los nervios que van del cerebro a la médula espinal) y el estudio de imágenes por resonancia magnética (para producir imágenes del interior del cuerpo). En esta revisión se describieron otros métodos utilizados actualmente como son las pruebas de análisis de sangre, orina, la biopsia de músculo o nervio y la evaluación de líquido cefalorraquídeo.
Así, el estudio del líquido cefalorraquídeo (LCR) sigue siendo de gran utilidad en el diagnóstico de enfermedades infecciosas, oncológicas y neurológicas. De hecho, se utiliza para diagnosticar el síndrome de Guillain Barré y la esclerosis lateral primaria, por lo que este tipo de análisis podría utilizarse para diagnosticar otras enfermedades motoras. Su extracción también sirve como medida terapéutica (administración de medicamentos). Varios de los componentes del líquido cefalorraquídeo son similares a los de la sangre, por lo que es recomendable comparar los valores obtenidos con los de una muestra de sangre obtenida de forma simultánea. Los valores normales de proteínas, glucosa y células (leucocitos) van a depender de la edad del paciente. Por lo que se debe considerar al líquido cefalorraquídeo como una vía de administración farmacológica, puesto que es una vía directa al sistema nervioso central tomando en cuenta que en algunas enfermedades su progresión es rápida, y también tomando en cuenta todas las limitantes que existen como el tamaño molecular, lipofilicidad, afinidad de los transportadores de la barrera hematoencefálica, entre otras.
PleasePor otro lado, se sabe que existe una relación entre las enfermedades motoras y las enfermedades cognitivas, ya que el desarrollo psicomotor en la primera infancia tiene un efecto predictor diferenciado sobre las alteraciones cognitivas en la edad escolar. Las dificultades que puede tener el niño de 0-6 meses en el desarrollo psicomotor y rehabilitado antes del año no tiene efecto sobre las alteraciones cognitivas del niño escolar. Sin embargo, si esas alteraciones persisten en la edad preescolar, entonces existe una posible predicción.69 Asimismo, se ha reportado que el deterioro cognitivo está presente en aproximadamente el 30% de los pacientes con ELA. Especialmente cuando esta patología es grave, ocurre un deterioro de la fluidez, la función ejecutiva, el lenguaje y la memoria.70 De esta manera, se deben de considerar tanto el diagnóstico motor y cognitivo de los pacientes.
Además, se ha demostrado que tener dificultades en el procesamiento sensorial puede afectar las habilidades motoras como mejorar la fuerza de los músculos de agarre de los miembros superiores, la coordinación, la velocidad de los movimientos, la destreza fina y bruta en los niños.74 También existen estudios que muestran que mucha de la sintomatología no motora suele estar asociada directamente a enfermedades motoras. Por ejemplo, en la enfermedad del Parkinson, estos síntomas incluyen: alteraciones de la memoria, dolor, depresión, ansiedad, alteraciones autonómicas (incontinencia urinaria, bradicardia, hipertensión), alteraciones gastrointestinales, alteraciones de sueño, etc.17 La duda que surge, es si estos síntomas podrían ser el origen de algunas enfermedades motoras raras como las descritas en el presente artículo.
Por otro lado, sujetos que padecen esclerosis lateral amiotrófica tienen afectaciones cognitivas como la velocidad de procesamiento de información, memoria de trabajo, memoria visual, verbal, lenguaje (fluencia verbal) y funciones ejecutivas. La prevalencia del deterioro cognitivo de pacientes con esclerosis múltiple fluctúa entre el 40 al 70%.1
De esta forma, también sería necesario realizar investigaciones analizando las afectaciones progresivas del procesamiento sensorial (como la propiocepción y el sistema vestibular) y de otras alteraciones (como las autonómicas y endocrinas) en sujetos que padecen enfermedades motoras.
10.1. Posibles blancos terapéuticos
Resulta conveniente obtener un mejor conocimiento de todos los factores genéticos y no genéticos involucrados en las enfermedades motoras para prevenirlas o para tener la esperanza de que algún día se consiga su cura absoluta en los sujetos que las padecen.
Existen terapias que controlan los síntomas o intentan retrasar la progresión de las enfermedades motoras. Uno de los tratamientos eficaces requiere restaurar la mielina perdida. La mielina es una capa aislante alrededor de los nervios que se desgasta gradualmente por la inflamación en la esclerosis múltiple y en otras enfermedades motoras. Los impulsos nerviosos que viajan por el cuerpo se ralentizan cuando está afectada esta capa, lo que provoca problemas neurológicos. Así, se ha reportado que la mielina está disminuida en una variedad de trastornos neurodegenerativos motores. De esta forma, habría que mejorar la producción de oligodendrocitos y, por lo tanto, estimular la reparación de la mielina. Las células madre producen una variedad de células cerebrales, incluidos los oligodendrocitos, un tipo de célula del sistema nervioso central que produce mielina. Con base en ello, un estudio realizado en ratones ha mostrado que una molécula inmunológica llamada fractalquina puede inducir en células madre la producción de oligodendrocitos que producen mielina, el cual es un factor clave en enfermedades como la esclerosis múltiple. Este tipo de terapias también podría aplicarse en otras enfermedades motoras.75
Adicionalmente, dada la evidencia de que el daño oxidativo por radicales libres ocurre como parte del proceso patogénico en la enfermedad de la neurona motora, sería un gran interés en probar tratamientos antioxidantes para analizar sus efectos en las enfermedades motoras.76 Algunos antioxidantes propuestos son las vitaminas C y E, la selegilina, el selenio, la metionina, la acetilcisteína y los curcuminoides.77-79
Finalmente, estudios recientes muestran que los videojuegos basados en tecnología de realidad virtual (es decir, Nintendo Wii, PlayStation Move y Kinect más XBOX 360) están emergiendo como herramientas válidas utilizadas en neurorrehabilitación para pacientes con esclerosis múltiple, las cuales mejoran la destreza manual brutal unilateral, la destreza manual fina y la coordinación. Esta tecnología podría utilizarse para la rehabilitación de otras enfermedades motoras, como las descritas en esta revisión.80 Se necesitan más investigaciones para explorar más a fondo diferentes nuevas pruebas de diagnóstico y de los posibles nuevos tratamientos e intervenciones apropiadas para combatir a las enfermedades motoras que hasta hoy en día son en su mayoría incurables.
Las neuronas motoras son células importantes en el sistema nervioso tienen diferentes funciones como integrar y transmitir información sensorial, motora y autonómica. Las afectaciones de las neuronas motoras forman parte de diferentes patologías motoras que en su mayoría son raras o de baja prevalencia encontradas en el mundo. Este tipo de neuronas se siguen estudiando para tener un mejor conocimiento de su etiología. Debido a la gravedad de los síntomas de estas enfermedades, la expectativa de vida es poca o nula en diferentes pacientes que pueden ser desde niños hasta adultos. Es necesario realizar más investigaciones para probar tratamientos farmacológicos o tratamientos alternativos contra las enfermedades motoras raras y así disminuir el índice de mortalidad en los sujetos que las padecen. No hay que olvidar que existen patologías prevenibles como en el caso del síndrome pospoliomielítico en donde la vacuna es un instrumento fundamental para evitar este tipo de patologías. En esta revisión se describieron algunas patologías motoras raras o poco comunes que pueden ser genéticas, virales o simplemente esporádicas. Nadie está exento de contraer cualquiera de éstas. Sin embargo, con el avance de la ciencia médica, se espera en un futuro próximo, encontrar tratamientos apropiados para obtener una mejor esperanza de vida en los sujetos que las padecen.
Los autores de este artículo de revisión han apoyado en su contenido y en la elaboración del formato de este escrito. Todos los autores declaran que no tienen conflicto de interés alguno.
Se agradece al Consejo Nacional de Ciencia y Tecnología (CONACyT) por la beca otorgada a A. J. L. F. (número 894595).
1. Custodio Bartol omé Herrera, N., nce Li ma, L., Custodio, N., Montesinos, R. & López-Góngora, M. Deterioro cognitivo en pacientes con esclerosis múltiple. An. la Fac. Med. (2018) 79:338–345.
2. Regensburger, M., Weidner, N. & Kohl, Z. Motor neuron diseases: Clinical and genetic differential diagnostics. Nervenarzt (2018) 89:658–665.
3. Héctor, M. R., Juan Didier, P.-G., María Elena, M., María Teresa, G.-G. & Jorge, M.-C. E. Esclerosis lateral amiotrófica. Contribución de la Neurología Mexicana de 1998 a 2014. Rev. Mex. Neurocienc. (2014) 15:355–362.
4. Pradat, P. F., Bernard, E., Corcia, P., Couratier, P., Jublanc, C., Querin, G., Morélot P. C., Salachas, F., Vial, C., Wahbi, K., Bede, P., Desnuelle, C., Le F. N., Echaniz-Laguna, A., Sorarù, G., Perez, T., Ramos, C., Goizet, C., Desport, J., Pugeat, M., Pichon, B., Maniez, S., Robillard, J., Coupe, C., Laurier, B. L., Roy, B. S., Lévêque, N., Penot, J., Goutines, C. V. The French national protocol for Kennedy’s disease (SBMA): Consensus diagnostic and management recommendations. Orphanet J. Rare Dis. (2020) 15:90.
5. El nombre y la prevalencia del Síndrome Postpolio - OMCETPAC. Available from: http://www.postpoliomexico.org/El_nombre_y_la_prevalencia_del_Sindrome_Postpolio.html (2013)
6. Orphanet: Esclerosis lateral primaria. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=35689 (2021)
7. En México se reconocen sólo 20 enfermedades raras, pero no a la Atrofia Muscular Espinal - LJA Aguascalientes. Available from: https://www.lja.mx/2020/03/en-mexico-se-reconocen-solo-20-enfermedades-raras-pero-no-a-la-atrofia-muscular-espinal/ (2020)
8. Rebolledo-García, D., González-Vargas, P. & Salgado-Calderón, I. Síndrome de Guillain-Barré: viejos y nuevos conceptos. Med. interna México(2018) 34:72–81.
9. La Esclerosis Lateral Amiotrófica ELA | Consejo Nacional para el Desarrollo y la Inclusión de las Personas con Discapacidad | Gobierno | gob.mx. Available from: https://www.gob.mx/conadis/articulos/la-esclerosis-lateral-amiotrofica-ela?idiom=es (2018)
10. Greco, A., Chiesa, M. R., Da Prato, I., Romanelli, A. M., Dolciotti, C., Cavallini, G., Masciandaro, S. M., Scilingo, E. P., Del Carratore, R., Bongioanni, P., Using blood data for the differential diagnosis and prognosis of motor neuron diseases: a new dataset for machine learning applications. Sci. Rep. (2021) 11.
11. Martínez, H. R., Escamilla-Ocañas, C. E. & Hernández-Torre, M. Non-motor neurological symptoms in patients with amyotrophic lateral sclerosis. Neurologia (2018) 33:474–476.
12. Riancho, J., Gonzalo, I., Ruiz-Soto, M. & Berciano, J. Why do motor neurons degenerate? Actualization in the pathogenesis of amyotrophic lateral sclerosis. Neurologia 1–11 (2016) 1:11.
13. Statland, J. M., Barohn, R. J., Dimachkie, M. M., Floeter, M. K. & Mitsumoto, H. Primary Lateral Sclerosis. Neurologic Clinics (2015) 33:749–760.
14. Bowerman, M., Murray, L. M., Scamps, F., Schneider, B. L., Kothary, R., Raoul, C. Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: Converging roads to therapeutic development. European Journal of Medical Genetics (2018) 61:685–698.
15. Rhodes, L. E., Freeman, B. K., Auh, S., Kokkinis, A. D., La Pean, A., Chen, C., Lehky, T. J., Shrader, J. A., Levy, E. W., Harris-Love, M., Di Prospero, N. A., Fischbeck, K. H. Clinical features of spinal and bulbar muscular atrophy. Brain (2009) 132:3242–3251.
16. Zapata-Zapata, Hugo, C., Franco-Dáger, ;, Solano-Atehortúa, E. ; & Marcos, J. Esclerosis lateral amiotrofica: actualización (2016) 29:8.
17. Mayo Clinic - Mayo Clinic. Available from: https://www.mayoclinic.org/. (2022).
18. Granatiero, V. & Manfredi, G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology (Basel) (2019) 8:36.
19. Singh, A., Kukreti, R., Saso, L. & Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules (2019) 24:1583.
20. Lanznaster, D., de Assis, D. R., Corcia, P., Pradat, P.-F. & Blasco, H. Metabolomics Biomarkers: A Strategy Toward Therapeutics Improvement in ALS. Front. Neurol. (2018) 9:1126.
21. Strohm, L. & Behrends, C. Glia-specific autophagy dysfunction in ALS. Semin. Cell Dev. Biol. (2019) 12:1061-1071.
22. Masrori, P. & Van Damme, P. Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. ene.14393 (2020) 99:172-182.
23. Fornes, P. claudia. Estudio sobre biomarcadores moleculares para el diagnóstico y el seguimiento de esclerosis lateral amiotrófica. Available from: https://riucv.ucv.es/handle/20.500.12466/445 (2019)
24. De Jong, S. W., Huisman, M. H. B., Sutedja, N. A., Van Der Kooi, A. J., De Visser, M., Schelhaas, H. J., Fischer, K., Veldink, J. H., Van Den Berg, L. H. Smoking, alcohol consumption, and the risk of amyotrophic lateral sclerosis: A population-based study. Am. J. Epidemiol. (2012) 176:233–239.
25. Torres Cuenca, T. F. Cuantificación de anormalidades electromiográficas en pacientes con esclerosis lateral amiotrófica. Available from: https://repositorio.unal.edu.co/handle/unal/79119 (2020)
26. Martínez, H., Escamilla-Ocañas, C., González-Garza, M. & Moreno Cuevas, J. Anormalidades clínicas y por resonancia magnética en lengua de pacientes con esclerosis lateral amiotrófica. Neurología (2018) 33:276–278.
27. David, D., Oramas, A., Javier, P. & Chinea, B. Esclerosis Lateral Amiotrófica y Cuidados de Enfermería: Revisión Bibliográfica Autora: Nacoremi Pérez Rodríguez. Available from: http://riull.ull.es/xmlui/handle/915/5360 (2017)
28. Domínguez, O. L., Ramos L. L. Toledo, L., Montes de Oca, T., Pérez, F., Spíritus-Cuba, S. Esclerosis lateral amiotrófica: un reto actual para las neurociencias Amyotrophic lateral sclerosis: a current challenge for neurosciences. (2018) 57:55–63.
29. García Narro, N. Relación de los niveles de vitamina D y su posible estrategia terapéutica en la enfermedad del ELA. Available from: http://hdl.handle.net/20.500.12466/1150. (2019)
30. Lezena Juberías, E. B., Martínez-González, L. & Martínez, A. Fármacos multidiana en el posible tratamiento de la esclerosis lateral amiotrófica. Available from: https://ebuah.uah.es/dspace/handle/10017/41480 (2019)
31. Gaitan, esteban sanchez & ampudia, margarita malpartida. revista medica sinergia. Rev. Medica Sinerg. (2021) 6:638-638.
32. Adriana Quintana, P. Esclerosis Lateral Amiotrófica (ELA): transformaciones que subyacen durante el proceso de rehabilitación. (2021) 100:1-21.
33. Riad, S. M., Hathout, H. & Huang, J. C. High T2 signal in primary lateral sclerosis supports the topographic distribution of fibers in the corpus callosum: Assessing 3disease in the primary motor segment. Am. J. Neuroradiol. (2011) 32:61-64.
34. Bede, P., Chipika, R. H., Finegan, E., Li Hi Shing, S., Doherty, M. A., Hengeveld, J. C., Vajda, A., Hutchinson, S., Donaghy, C., McLaughlin, R. L., Hardiman, O. Brainstem pathology in amyotrophic lateral sclerosis and primary lateral sclerosis: A longitudinal neuroimaging study. NeuroImage Clin. (2019) 29:105229.
35. Yedavalli, V. S., Patil, A. & Shah, P. Amyotrophic lateral sclerosis and its mimics/variants: A comprehensive review. Journal of Clinical Imaging Science (2018) 8:53.
36. Martinez Martinez, M. & Rodriguez Leyva, I. Esclerosis lateral primaria (reporte de un caso clínico y revisión de las manifestaciones clínicas). Available from: http://previous.revmexneurociencia.com/wp-content/uploads/2014/07/Nm0013-05.pdf (2001)
37. Esclerosis lateral primaria (PLS) // Middlesex Health. Available from: https://middlesexhealth.org/learning-center/espanol/enfermedades-y-afecciones/esclerosis-lateral-primaria-elp (2021)
38. Kolb, S. J. & Kissel, J. T. Spinal Muscular Atrophy. Neurologic Clinics (2015) 33:831–846.
39. Orphanet: Atrofia muscular espinal proximal tipo 2. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=83418. (2022)
40. Informe de Posicionamiento Terapéutico de nusinersen (Spinraza®) en atrofia muscular espinal. Available from: https://www.fundame.net/investigacion-ame/ultimas-noticias/421-informe-de-posicionamiento-terapeutico-de-nusinersen-spinraza-en-atrofia-muscular-espinal.html (2015)
41. Haaker, G. & Fujak, A. Proximal spinal muscular atrophy: Current orthopedic perspective. Application of Clinical Genetics (2013) 6:113–120.
42. Atrofia muscular espinal | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Available from: https://rarediseases.info.nih.gov/espanol/11864/atrofia-muscular-espinal (2017)
43. Ferrari, E. T. ATROFIA MUSCULAR ESPINAL INFANTIL. Boletín del Real Patronato sobre Discapacidad. (2007) 62:6-15.
44. Lorson, C. L., Rindt, H. & Shababi, M. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum. Mol. Genet. (2010) 19:11-118.
45. Coady, T. H. & Lorson, C. L. Trans-Splicing-Mediated Improvement in a Severe Mouse Model of Spinal Muscular Atrophy. J. Neurosci. (2010) 30:126.
46. Williams, J. H., Schray, R. C., Patterson, C. A., Ayitey, S. O., Tallent, M. K., Lutz, G. J. Oligonucleotide-Mediated Survival of Motor Neuron Protein Expression in CNS Improves Phenotype in a Mouse Model of Spinal Muscular Atrophy. J. Neurosci. (2009) 29:7633.
47. Hua, Y., Sahashi, K., Hung, G., Rigo, F., Passini, M. A., Bennett, C. F., Krainer, A. R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. (2010) 24:1634.
48. Giménez, G., Bersano, C., Herrero, M., Manresa, A., Pronello, D., Salinas, P. Recomendaciones para el manejo respiratorio de los pacientes con atrofia muscular espinal Recommendations for respiratory management of patients with spinal muscular atrophia Recomendações para a manipulação respiratória de pacientes com atrofia muscular espinhal. (2021) 9:401.
49. Sturm, S., Günther, A., Jaber, B., Jordan, P., Kotbi, N., Parkar, N., Cleary, Y., Frances, N., Bergauer, T., Heinig, K., Kletzl, H., Marquet, A., Ratni, H., Poirier, A., Müller, L., Czech, C., Khwaja, O. A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br. J. Clin. Pharmacol. (2019) 85:181.
50. DailyMed - ZOLGENSMA- onasemnogene abeparvovec-xioi kit. Available from: https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=68cd4f06-70e1-40d8-bedb-609ec0afa471# (2021).
51. Arméstar, F., Arméstar, F., Catalán, B. & Martínez, S. Síndrome de Guillain Barré en la Unidad de Cuidados Intensivos. Rev. Médica Trujillo (2018) 13:100-102.
52. Silva, M. A., Palacios, E., Castillo, G. A., Monsalve, J. A. & Leal Castaño, L. F. Secuelas neurológicas del síndrome de Guillain-Barré en pacientes adultos. Rev. Repert. Med. y Cirugía (2020) 29:185–191.
53. Leonhard, E., Mandarakas, M., Gondim, A., Bateman, K., Ferreira, M., Cornblath, D., Van Doorn, P., Dourado, M., Hughes, R., Islam, B., Kusunoki, S., Pardo, C., Reisin, R., Sejvar, J., Shahrizaila, N., Soares, C., Umapathi, T., Wang, Y., Yiu, E., Willison, H., Jacobs, B. Diagnosis and management of Guillain–Barré syndrome in ten steps. Nat. Rev. Neurol. (2019) 15:671–683.
54. Esposito, S. & Longo, M. R. Guillain–Barré syndrome. Autoimmun. Rev. (2017) 16:96–101.
55. Pérez-Lledó, E., Díaz-Vico, A. & Gómez-Gosálvez, F. A. Síndrome de Guillain-Barré: presentación clínica y evolución en menores de 6 años de edad. An. Pediatría (2012) 76:69–76.
56. Quilismal Guanochanga, N. C., Puglla Sanchez, L. R., Parra Paladines, W. G., Pérez López, J. S. & Pillajo Gavilanes, K. E. Síndrome de Guillain Barré en paciente con antecedente de infección respiratoria: reporte de un caso. Mediciencias UTA (2020) 4:46.
57. Chang-Quezada, S. & Hernández, E. Síndrome Guillain-Barré en pacientes con antecedentes de virus epidémicos de Zika y SARS-CoV-2. Una revisión narrativa. Ciencia, Tecnología y Salud (2020) 7:396-409.
58. Valera Yepes, R., Virgili Casas, M., Povedano Panades, M., Guerrero Gual, M. & Villabona Artero, C. Enfermedad de Kennedy y resistencia parcial androgénica. Descripción de 4 casos y revisión de la literatura. Endocrinol. y Nutr. (2015) 62:224–230.
59. Grunseich, C., Rinaldi, C. & Fischbeck, K. Spinal and bulbar muscular atrophy: Pathogenesis and clinical management. Oral Dis. (2014) 20:6–9.
60. Manzano, R., Sorarú, G., Grunseich, C., Fratta, P., Zuccaro, E., Pennuto, M., Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. Journal of Neurology, Neurosurgery and Psychiatry (2018) 89:808–812.
61. Pennuto, M. & Rinaldi, C. From gene to therapy in spinal and bulbar muscular atrophy: Are we there yet? Molecular and Cellular Endocrinology (2018) 465:113–121.
62. Moraes, M., Calvo, J., Montano, N., Rodríguez, X., Quadrelli, R., Vaglio, A., Atrofia muscular espinal y bulbar: enfermedad de Kennedy. Aspectos clínicos y genéticos. Revista Médica Del Uruguay. (2019) 35:160–175.
63. Durán-Salgado, M. B., Fernández-Valverde, F., Vargas-Cañas, E., Medina-Luna, P. & Velasco Suárez, M. Enfermedad de Kennedy (atrofia muscular espino-bulbar). Reporte de caso y revisión de la bibliografía. (2013) 29:213-218.
64. Rocchi, A. & Pennuto, M. New routes to therapy for spinal and bulbar muscular atrophy. J. Mol. Neurosci. (2013) 50:514–523.
65. Saraswathy, R. The genetics of post-polio syndrome - Much to be unravelled! Indian Journal of Medical Research (2016) 144:9–10.
66. Boyer, F. C., Tiffreau, V., Rapin, A., Laffont, I., Percebois-Macadré, L., Supper, C., Novella, J. L., Yelnik, A. P. Post-polio syndrome: Pathophysiological hypotheses, diagnosis criteria, medication therapeutics. Annals of Physical and Rehabilitation Medicine (2010) 53:34–41.
67. Baj, A., Colombo, M., Headley, J. L., McFarlane, J. R., Liethof, M., Toniolo, A. Post-poliomyelitis syndrome as a possible viral disease. International Journal of Infectious Diseases (2015) 35:107-116.
68. Poliomielitis - OPS/OMS | Organización Panamericana de la Salud. https://www.paho.org/es/temas/poliomielitis#:~:text=La%20poliomielitis%20puede%20ocasionar%20par%C3%A1lisis,y%20nuevos%20episodios%20de%20par%C3%A1lisis. (2021)
69. Sáinz, M. P., Pelayo, R., Laxe, S., Castaño, B., Capdevilla, E., Portell, E. Describing post-polio syndrome. Neurologia (2019) 9:1312.
70. Saiz Echezarreta, M. Poliomielitis y síndrome postpolio. Toda una vida superando obstáculos INVESTIGACIÓN CUALITATIVA. Nuberos Científica vol. 2 www.enfermeriadecantabria.com/nuberoscientica (2015) 11:3.
71. Willen, C., Hou, L. & Sunnerhagen, K. S. A very long term longitudinal follow up of persons with late effects of polio. Eur. J. Phys. Rehabil. Med. (2020) 56:155-159.
72. Davies, R. A., Maher, C. G. & Hancock, M. J. A systematic review of paracetamol for non-specific low back pain. Eur. Spine J. (2008) 17:1423.
73. Moore, R. A., Derry, S., Straube, S., Ireson-Paine, J. & Wiffen, P. J. Faster, higher, stronger? Evidence for formulation and efficacy for ibuprofen in acute pain. Pain (2014) 155:14–21
74. Yeger, B. E. Sensory processing difficulties (SPD) and their relation to motor performance and to child’s perceived competence among children with developmental coordination disorders. Int. Phys. Med. Rehabil. J. (2018) 3:120–126.
75. Watson, A., De Almeida M., Dittmann, N., Li, Y., Torabi, P., Footz, T., Vetere, G., Galleguillos, D., Sipione, S., Cardona, A., Voronova, A. Fractalkine signaling regulates oligodendroglial cell genesis from SVZ precursor cells. Stem cell reports (2021) 16:1968–1984.
76. Talebi, M., Farkhondeh, T., Kopustinskiene, D., Simal-Gandara, J., Bernatoniene, J., Samarghandian, S. An updated review on the versatile role of chrysin in neurological diseases: Chemistry, pharmacology, and drug delivery approaches. Biomed. Pharmacother. (2021) 141:111906.
77. Silvestro, S., Sindona, C., Bramanti, P. & Mazzon, E. A State of the Art of Antioxidant Properties of Curcuminoids in Neurodegenerative Diseases. Int. J. Mol. Sci. (2021) 22:1–26.
78. Sies, H. & Stahl, W. Vitamins E and C, beta-carotene, and other carotenoids as antioxidants. Am. J. Clin. Nutr. (1995) 62:1315-1321.
79. Duarte-Jurado, A., Gopar-Cuevas, Y., Saucedo-Cardenas, O., Loera-Arias, M., Montes-De-oca-luna, R., Garcia-Garcia, A., Rodriguez-Rocha, H. Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities. Antioxidants (Basel, Switzerland) (2021) 10:1–19.
80. Cuesta-Gómez, A., Sánchez-Herrera-Baeza, P., Oña-Simbaña, E., Martínez-Medina, A., Ortiz-Comino, C., Balaguer-Bernaldo-de-Quirós, C., Jardón-Huete, A., Cano-de-la-Cuerda, R. Effects of virtual reality associated with serious games for upper limb rehabilitation inpatients with multiple sclerosis: randomized controlled trial. J. Neuroeng. Rehabil. (2020) 17:90.
| Recibido: 21 de octubre, 2021 | Aceptado: 26 de febrero, 2022 |
*Correspondencia: Dr. César Antonio Pérez Estudillo. Instituto de Investigaciones Cerebrales, Universidad Veracruzana. Av. Luis Castelazo S/N Col. Industrial Ánimas Xalapa, Ver. C.P. 91190 Teléfono: 52(228)8418900 ext. 13059. E-mail: cesperez@uv.mx
Este es un artículo de libre acceso distribuido bajo los términos de la licencia de Creative Commons, (http://creamasal@unam.mxtivecommons.org/licenses/by-nc/3.0), que permite el uso no comercial, distribución y reproducción en algún medio, siempre que la obra original sea debidamente citada.