El transporte de la glucosa en las células gliales del cerebro obeso
- Inicio
- Comité Editorial
- Lineamientos
- Carta de Cesión de Derechos
- Información Legal
- Acerca de la Revista
- Bases de Datos
- Contacto
- ISSN 2007-3054
- Centro de Investigaciones Cerebrales
Universidad Veracruzana
López-Ramírez Gabriel, Méndez-Flores Orquidia G*
Universidad Juárez Autónoma de Tabasco, División Académica de Ciencias de la Salud, Villahermosa, Tabasco, México. CP. 86690.
Resumen
Abstract
Introducción
El cerebro obeso
Células gliales del cerebro obeso y la señalización de la leptina
Transportadores gliales de glucosa
Transporte de glucosa en el cerebro obeso
Conclusión
Conflicto de intereses
Financiamiento
Referencias
Mail
La pandemia de enfermedades metabólicas, asociada a la transición nutricional reciente y al estilo de vida sedentario, afecta la salud del individuo y modifica la funcionalidad del cerebro. La inflamación de bajo grado generalizada (desregulación inmunológica crónica y sistémica, asociada al tejido adiposo) durante las afecciones metabólicas desencadena neurodegeneración, envejecimiento del cerebro y deterioro cognitivo. Estos cambios funcionales están correlacionados con una tasa metabólica baja, con pérdida neuronal y con una gliosis reactiva. El objetivo del presente trabajo fue condensar y analizar la información relacionada con la participación del transporte de glucosa de las estirpes gliales, durante el mantenimiento de la homeostasis metabólica en el cerebro obeso. Para este fin, revisamos la base de datos PubMed y reunimos la información más reciente al respecto. Concluimos que las citocinas pro-inflamatorias y los mediadores químicos de la alimentación intervienen en la modulación de los principales transportadores de glucosa (GLUT1-GLUT5) en las células gliales del cerebro, además de que se relacionan con la disminución en la expresión del transportador mayoritario de la barrera hematoencefálica, GLUT1, y la condición de inanición en el cerebro. Los mecanismos moleculares implicados en la obesidad comparten un gran parecido con los cambios metabólicos en sobrepeso y en condiciones de malnutrición por exceso. Estas desregulaciones tempranas están vinculadas a la tasa metabólica baja del encéfalo, la neurodegeneración y también puede explicar los cambios identificados en el comportamiento y en la cognición, los cuales prevalecen en un cerebro obeso.
Palabras clave: transporte de glucosa, obesidad, metabolismo, células gliales.
The pandemic of metabolic diseases, associated with the recent nutritional transition and sedentary lifestyle, affects the health of the individual and modifies the functionality of the brain. Widespread low-grade inflammation (chronic and systemic immune dysregulation, associated with adipose tissue) during metabolic conditions triggers neurodegeneration, brain aging, and cognitive decline. These functional changes are correlated with low metabolic rate, neuronal loss, and reactive gliosis. The objective of the present work was to condense and analyze the information regarding the participation of glucose transport of glial lineages, during the maintenance of metabolic homeostasis in the obese brain. For this purpose, we reviewed the PubMed database and gathered the most recent information on it. We conclude that pro-inflammatory cytokines and chemical mediators of food are involved in the modulation of the main glucose transporters (GLUT1-GLUT5) in the glial cells of the brain, in addition to being related to the decrease in the expression of the main transporter of the blood-brain barrier, GLUT1, and the starvation condition in the brain. The molecular mechanisms involved in obesity share a great resemblance to the metabolic changes in overweight and under conditions of excess malnutrition. These early dysregulations are linked to the brain's low metabolic rate, neurodegeneration, and may also explain the identified changes in behavior and cognition that are associated with the obese brain.
Keywords: glucose transport, obesity, metabolism, glial cells.
La transición nutricional y epidemiológica reciente ha favorecido la prevalencia de los padecimientos metabólicos. Estos cambios están asociados a la estandarización de la dieta a nivel mundial, donde las demandas crecientes de alimentos por parte de la población humana incentivan la producción masiva y simplificada de recursos alimentarios industrializados. Por lo tanto, la oferta alimentaria está dominada por productos procesados, ricos en carbohidratos refinados, grasas saturadas, enriquecimiento nutricional artificial, pero con carencia de agua y fibra dietética. Por otro lado, las adaptaciones dietéticas en el ajuste equilibrado del tamaño de las porciones y el comportamiento alimentario en general, conforme a los requerimientos nutricionales, la suplementación con micronutrientes deficientes y las adecuaciones en el estilo de vida están rezagadas en su aplicación; por lo tanto, las afectaciones metabólicas y sus efectos concomitantes en la salud son el común denominador en las poblaciones actuales.1,2
Las repercusiones a nivel sistémico de los padecimientos metabólicos son diversas. El abanico de afectaciones va desde el sobrepeso hasta la obesidad e involucra desregulaciones en la tolerancia a la insulina, a la leptina, desarrollo de diabetes mellitus, hipertensión arterial sistémica, enfermedades cardiovasculares, disfunciones endocrinológicas, esteatosis hepática, afectaciones reproductivas, inmunológicas y cáncer.3,4 La trascendencia epidemiológica de esta prevalencia alta de padecimientos metabólicos se extiende hasta las tasas de productividad, donde puede ser documentado en términos de años de vida productiva, índices de calidad de vida, morbilidad y mortalidad. Estos últimos indicadores son de especial atención porque sobrecargan los sistemas de atención a la salud, los mecanismos y recursos para la distribución de la misma,5 sobre todo considerando las afectaciones neurológicas que se asocian a los padecimientos metabólicos.
Actualmente, existen innumerables investigaciones epidemiológicas de afecciones metabólicas y de los mecanismos moleculares involucrados en esta condición patológica; en particular acerca del transporte de la glucosa en el sistema nervioso tomando como principal protagonista a las neuronas, en condiciones muy diversas (modelos biológicos, salud y enfermedad), pero pocos estudios en las células gliales. Es a partir de los últimos 10 años cuando se duplicó la cantidad de artículos publicados en esta área del conocimiento. Por lo tanto, el objetivo del presente trabajo fue describir el papel que desempeñan las células gliales en condiciones fisiológicas y de obesidad, desde el aspecto del transporte de la glucosa en las células gliales y el desequilibrio metabólico que genera en el organismo. Esto se asocia a que existe un desequilibrio en la señalización basal del transporte de la glucosa en condiciones de obesidad con respecto de las fisiológicas, en las proteínas transportadoras de glucosa que se expresan en los distintos tipos gliales del cerebro. Para tal efecto, realizamos una búsqueda sistemática de artículos recientes que fueron encontrados a través de los criterios siguientes en la base de datos PubMed, del “National Library of Medicine, National Center for Biotechnology Information”: [glial cells] [obesity] [glucose transport]. Concluimos que el sistema de transporte de glucosa en las células gliales tiene una participación preponderante, ya que existe una alteración en los mecanismos funcionales desde el inicio del consumo de las dietas hipercalóricas y que estos cambios, están relacionados a una inflamación crónica de bajo grado y a hormonas moduladoras del apetito en el cerebro obeso.
El sistema nervioso central (SNC) está conformado por dos tipos de células: las células neuronales y las células gliales. Estás ultimas se clasifican de acuerdo con su función en: oligodendrocitos, astrocitos y microglía. El mantenimiento de la función cognitiva normal es un proceso complejo que involucra la coordinación de la función neuronal con las glías del cerebro. El equilibrio de la bioenergética es un requisito central de las neuronas para realizar sus funciones, ya que necesitan grandes cantidades de energía, pero no pueden almacenarla en su forma convencional de glucógeno. Por lo tanto, las células gliales son necesarias para brindar sustento metabólico, mismo que realizan gracias a que expresan una amplia variedad de transportadores de glucosa, y de esta forma aseguran un suministro energético adecuado para las neuronas.6
El desequilibrio de la bioenergética ocasionado por la obesidad y las enfermedades metabólicas desregulan la función glial, lo que lleva a una falla para poder responder a las demandas de energía de las neuronas, generando así daño neuronal. En este mismo sentido las células gliales también participan como mediadores críticos de la respuesta a la inflamación en el cerebro. La diversidad de funciones que se han demostrado a las glías, son gracias a sus características que incluyen la flexibilidad metabólica, la sensibilidad a los cambios en el microambiente del SNC y la capacidad de adaptar rápidamente su función bajo demanda. Por lo tanto, son células especializadas que cooperan para promover y preservar la salud neuronal, desempeñando un papel importante en la regulación de la actividad de las redes neuronales en todo el cerebro. La creciente evidencia apunta a un papel fundamental de la glía, más notablemente en los tipos de astrocitos y la microglía, en la regulación sistémica de la homeostasis de la energía y la glucosa en el curso del control fisiológico normal y durante la enfermedad.6,7
El sistema nervioso (SN) también es afectado por los desórdenes metabólicos. Algunos ejemplos de patologías son las mielopatías metabólicas, las neuropatías periféricas, los síntomas cognitivos y psiquiátricos.8 El cerebro obeso tiene como características principales la inflamación y la senescencia. El sobrepeso y la obesidad, a nivel sistémico, son condiciones metabólicas de exceso de adiposidad que suponen riesgos importantes para la salud del individuo. El parámetro índice de masa corporal es el exceso de peso por área corporal (IMC), ya sea igual o mayor a 25 kg/m2 para sobrepeso o igual / mayor a 30 kg/m2 para la obesidad.9,10 En el cerebro también se han identificado pautas características del sobrepeso, obesidad e hiperfagia, que están asociadas con acciones específicas del sistema inmunológico y en el ciclo de vida celular. La adiposidad, puede producir citocinas como IL-1b (interleucina 1-beta), IL6 (interleucina 6), IFNγ (interferón gamma), TNFα (Tumor Necrosis Alpha, factor de necrosis tumoral-alfa) y MCP1(Monocyte Chemoattractant protein-1, proteína quimioatrayente de monocitos 1); estas moléculas señalizadoras activan a NF-kb (Nuclear factor kappa-light-chain-enhancer of activated B cells, Factor nuclear potenciador de las cadenas ligeras kappa de células B activadas) y promueven un estado de inflamación crónica de bajo grado. Esta inflamación (la activación del complejo enzimático inflamasoma por sustancias endógenas, como cristales de colesterol dentro de los macrófagos en arteriosclerosis, así como por ácidos grasos libres y lípidos en tejido adiposo, conduce a la liberación de IL-1b e IL-18) se extiende al cerebro debido a la disfunción endotelial que se desarrolla en la barrera hematoencefálica (BHE), de modo que los signos identificables son el estrés oxidativo, la inducción de muerte celular y el deterioro cognitivo.11 La pérdida de la capacidad proliferativa, la secreción de proteínas pro-inflamatorias (IL-1b, IL6, IL8) y de factores de crecimiento característicos de senescencia - NF-kb - (“Senescence- Associated Secretory Phenotype, SASP”) definen el envejecimiento celular, el cual ha sido encontrado en células gliales, ya que son las células del SN que pueden proliferar.12–14
La reducción del volumen de la materia gris que ha sido reportada en seres humanos y en modelos biológicos de sobrepeso y obesidad puede explicarse por la pérdida de poblaciones neuronales. Se desconocen los mecanismos de reducción del volumen de la materia gris, sin embargo, es probable que la neuroinflamación causada por la obesidad induzca la pérdida neuronal. Los adipocitos, los macrófagos del tejido adiposo y la disbiosis intestinal en individuos obesos y con sobrepeso dan como resultado la secreción de citocinas (TNF-α, IL-6, y IL-12) y quimiocinas (lipopolisacáridos y IFN-γ) que cruzan la barrera hematoencefálica y pueden estimular a la microglía, que a su vez también libera citocinas proinflamatorias (IL-1β, IL-6, TNF-α y INF-γ) dentro del cerebro. Esto conduce a una neuroinflamación crónica de bajo grado que puede señalizar para apoptosis y muerte neuronal. Además, la microangiopatía significativa observada en modelos de rata puede ser otro mecanismo importante de inducción de apoptosis.15
La función cerebral en modelos de obesidad y en humanos se ha mostrado afectada en aspectos del control del comportamiento (funciones ejecutivas), el aprendizaje y la memoria.16 Las neuronas hipotalámicas son de constante abordaje en estos estudios, ya que fungen como integradoras de información sobre el almacenamiento de energía a largo plazo, la disponibilidad de nutrientes a corto plazo y la demanda metabólica.17 Sin embargo, recientemente, las células gliales han ganado especial atención por su estrecha participación en la neuroinflamación y los trastornos metabólicos, como la obesidad y la diabetes.
3. Células gliales del cerebro obeso y la señalización de la leptina
En el cerebro sano, se producen interacciones metabólicas entre las glías y las neuronas cuidadosamente organizadas para mantener el funcionamiento normal del cerebro.6 El síndrome metabólico, la obesidad y la diabetes generan una interrupción glial, lo cual desencadena perturbaciones en las interacciones glía-neurona con una pérdida de transferencia de sustrato de energía a las neuronas que necesitan apoyo metabólico. En última instancia, esto conduciría al fallo de la bioenergética neuronal y la transmisión de señales y, finalmente, a la pérdida neuronal y al deterioro cognitivo.
Con respecto a los astrocitos, las evidencias respaldan el supuesto de que el desequilibrio metabólico entre los astrocitos y las neuronas promueven el deterioro cognitivo en la obesidad y el síndrome metabólico. Múltiples estudios preclínicos y clínicos también confirman que la obesidad, la diabetes y los componentes del síndrome metabólico promueven el deterioro cognitivo y que los cambios en las interacciones metabólicas de las neuronas y los astrocitos son fundamentales en enfermedades neurodegenerativas.18
La influencia del consumo de dieta alta en grasa y la obesidad en la comunicación microglía-neurona en las regiones cerebrales son responsables de la función cognitiva y la memoria. En el hipocampo, una estructura límbica que contribuye a la memoria y las tareas de aprendizaje muestra activación microglial en modelos de ratones con obesidad inducida por la dieta.19,20 La obesidad se asocia con deterioro cognitivo en el hipocampo en modelos de roedores.21
Las células gliales contribuyen a la regulación del balance energético y la patogénesis de la obesidad. Comenzando con los primeros trabajos sobre la señalización de la leptina en los astrocitos, esta área de investigación surgió rápidamente después del descubrimiento de la inflamación hipotalámica y la gliosis reactiva en modelos de roedores y en humanos obesos. Los estudios actuales han revelado la participación de una variedad amplia de tipos de células gliales en la modulación de la actividad neuronal, la regulación de la disponibilidad de hormonas, nutrientes, y la participación en la regulación fisiológica del comportamiento alimentario. Además, recientemente se ha implicado a un tipo glial, la microglía, en la susceptibilidad a la obesidad inducida por la dieta.17 Además, se ha evidenciado que varios moduladores endocrinos, incluidos la leptina y la grelina, tienen funciones neuroprotectoras en enfermedades neurológicas. Los receptores de leptina en el hipotálamo son el objetivo principal de la regulación homeostática del peso corporal. Trabajos recientes muestran que los receptores de leptina también se expresan en otras regiones del SNC, como el hipocampo, la corteza cerebral y la médula espinal. En consecuencia, estos estudios han identificado la participación de la leptina en la regulación de la supervivencia y el desarrollo neuronal. Además, se ha demostrado que la leptina tiene funciones neuroprotectoras en modelos animales de enfermedades neurológicas y de desmielinización. Estas observaciones también sugieren que la desregulación de la señalización de la leptina puede estar involucrada en la asociación entre la neurodegeneración y la obesidad.22 Se considera que la comunicación glía-glía es un mecanismo fundamental de la inflamación hipotalámica crónica inducida por la malnutrición por exceso, que podría estar intrínsecamente asociada a la obesidad, la diabetes y a sus demás consecuencias patológicas.23
4. Transportadores gliales de glucosa
Los transportadores de glucosa son proteínas integrales de membrana que permiten el transporte de glucosa y de sustancias estructuralmente relacionadas a través de las membranas celulares. Se han identificado dos familias de transportadores de glucosa: a) la familia de transportadores de glucosa de difusión facilitada (símbolo del gen Slc2a, símbolo de la proteína GLUT, de “Glucose Transporter”), la cual se constituye por trece miembros (GLUT 1-12, HMIT y 4 pseudogenes); b) la familia de transportadores dependientes de Na+ (símbolo del gen Slc5a, símbolo de la proteína SGLT), de la que se han reportado seis miembros, pero solo SGLT1 y SGLT2 han sido bien caracterizados en el encéfalo.24,25
En el cerebro, el GLUT1 es el que tiene una mayor expresión en células gliales. Esta proteína se configura en dos isoformas codificadas por el mismo gen y que difieren únicamente en su extensión de glucosilación. La isoforma de 55 kDa se encuentra localizada en la membrana luminal y abluminal de las células endoteliales de los capilares del cerebro presentes en la BHE; mientras que la isoforma GLUT1 de 45 kDa se expresa en todas las células neuronales, pero mayormente en astrocitos y oligodendrocitos, pero no así en la microglía.26,27 Por su parte, las células microgliales, que son de origen hematopoyético al ser macrófagos residentes en el cerebro, expresan mayoritariamente la isoforma GLUT5.28
5. Transporte de glucosa en el cerebro obeso
Como resultado de la inflamación crónica de grado bajo que se asocia al sobrepeso, la obesidad o a las dietas hipercalóricas, el sistema inmunológico juega un papel preponderante en la regulación del transporte de la glucosa. De tal suerte que los incrementos locales y súbitos en las concentraciones de glucosa, lo cual es de esperarse en la región proximal a la BHE, se asocia con aumentos en la función y activación de la microglía, favoreciendo así su transporte de glucosa.29 Por otro lado, la producción de interleucinas pro-inflamatorias como IL-1B regula positivamente la expresión de GLUT1 en el endotelio y en astrocitos.30
La alimentación está regulada por un mecanismo de control hedónico y otro homeostático. En el ámbito de la regulación homeostática las hormonas grelina y leptina, con características orexigénicas y anorexigénicas, respectivamente, toman relevancia. Las células gliales constan de receptores a leptina, sobre todo en la región hipotalámica asociada a la percepción de los nutrientes; por lo tanto, es de esperarse una regulación sobre los transportadores de glucosa, GLUT2 principalmente. En el caso del transportador GLUT1, puede observarse una regulación compleja, ya que puede aumentar su expresión en etapas tempranas de nutrición por exceso, lo cual podría coincidir con señales de leptina en el parénquima cerebral, pero puede disminuir los niveles de este transportador en la BHE como mecanismo de protección frente a la hiperglucemia sistémica y también puede responder a concentraciones de insulina a través de proteínas de andamiaje que facilitan su anclaje a la membrana y limitan su funcionalidad. Por su parte, el GLUT3 disminuye su expresión cuando existen condiciones que predisponen a la ganancia de peso corporal, lo cual se correlaciona con la disminución en el volumen de la materia. Mayores detalles pueden constatarse con la evidencia mostrada en la tabla 1.29,31–38
El cerebro debe mantener una concentración constante de glucosa; sin embargo, existen rangos de tolerancia. La glucosa en condiciones euglucémicas es de aproximadamente 1 ± 0.1 mmol/ml (equivalente a 4.7 mM de concentración plasmática), pero en hiperglucemia se ha encontrado valores de 1.8 a 2.7 mmol/ml (equivalente a 7.3 y 12.1 mM de concentración plasmática).39 Los transportadores de glucosa, sobre todo los que se encuentran en la BHE deben ser regulados conforme a las necesidades. Por lo tanto, GLUT1, transportador mayoritario de las células gliales astrocíticas y endoteliales de los vasos sanguíneos, se ha encontrado regulado negativamente en condiciones de dietas hipercalóricas, en diabetes y en la enfermedad de Alzheimer (enfermedad metabólica cuando es de inicio tardío), sugiriendo así un mecanismo de protección contra las hiperglucemia sistémica imperante, poniendo en déficit de glucosa al cerebro.36,40,41 La regulación del mismo transportador puede estar asociada a mensajeros químicos metabólicos o a fracciones proteicas de desecho que se acumulan en el caso de la neurodegeneración.
Los transportadores de glucosa distintos a GLUT1 también son afectados por las condiciones metabólicas del organismo. El transportador GLUT2 se ha visto asociado a la percepción de los niveles de glucosa en el torrente sanguíneo desde los tanicitos. En el caso de GLUT5 este puede ser regulado por mediadores de la inflamación de bajo grado. El transportador de glucosa en neuronas por excelencia, el GLUT3, presenta niveles de expresión ajustables a la disponibilidad de nutrientes, ya que en condiciones de malnutrición por exceso o en presencia de leptina disminuye, pero aumenta cuando hay ayuno. Todos los mecanismos aquí expuestos demuestran que la regulación negativa de los transportadores de glucosa en el cerebro explicaría la baja actividad metabólica del mismo en condiciones de sobrepeso u obesidad, sobre todo considerando que las células gliales son los elementos celulares predominantes que expresan los transportadores de glucosa más importantes en el SNC (GLUT1, GLUT2, GLUT3, GLUT4 y GLUT5). Finalmente, el cerebro obeso además de mostrar características de inflamación crónica subclínica o de bajo grado, de senescencia por la muerte neuronal y perfil SASP en sus células gliales, también presenta una disminución en la expresión de GLUT1 en la BHE, un aumento de GLUT5 por la activación microglial y una disminución de la expresión de GLUT3 por la disminución de la materia gris.

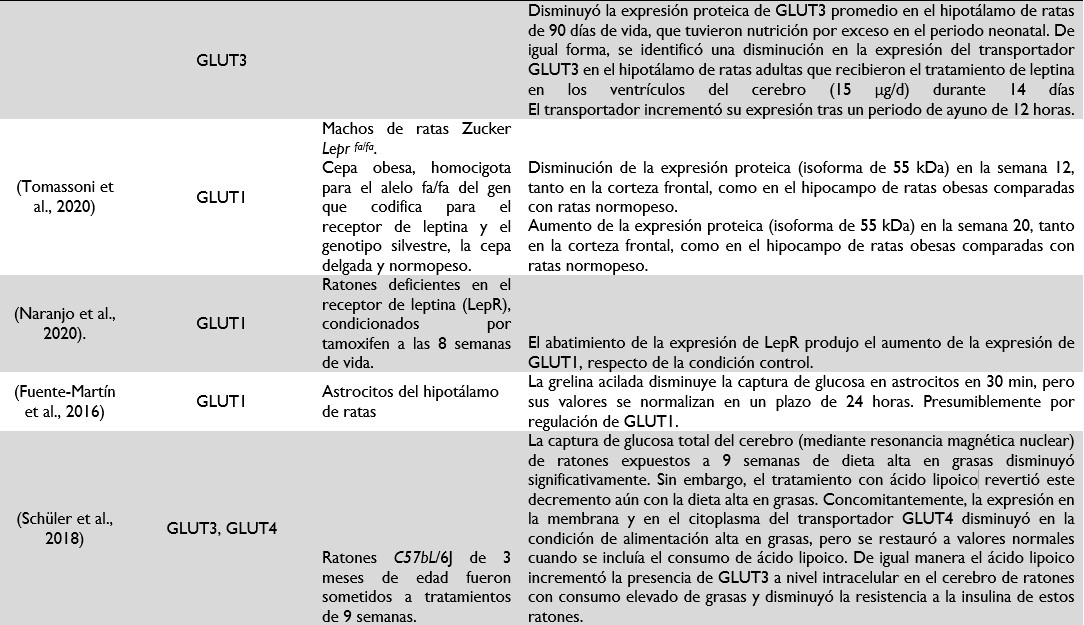

Tabla 1: Trabajos recientes que describen el transporte de la glucosa en el cerebro obeso. Se describe de forma abreviada los abordajes metodológicos y principales descubrimientos reportados en las publicaciones más recientes en el área de los transportadores de glucosa gliales en condiciones de desregulación metabólica.
Los autores de este artículo de revisión han contribuido en el diseño y en el contenido de este escrito, así mismo, se declara que no tenemos ninguna situación de conflicto de interés real, potencial o evidente, en relación con la presente obra titulada: “El transporte de la glucosa en las células gliales del cerebro obeso”.
La elaboración del trabajo aquí presentado fue independiente de patrocinio o beca alguna.
- Rivera JA, Barquera S, González-Cossío T, Olaiz G, Sepúlveda J. Nutrition Transition in Mexico and in Other Latin American Countries. Nutr Rev. 2004;62:S149-S157. doi:10.1111/j.1753-4887.2004.tb00086.x
- Ortega MA, Fraile-Martínez Ó, García-Montero C, Alvarez-Mon MA, Lahera G, Monserrat J, Llavero-Valero M, Mora F, Rodríguez-Jiménez R, Fernandez-Rojo S, Quintero J, Alvarez De Mon M. Nutrition, Epigenetics, and Major Depressive Disorder: Understanding the Connection. Front Nutr. 2022;9:867150. doi:10.3389/fnut.2022.867150
- Ríos-Hoyo A, Cortés MJ, Ríos-Ontiveros H, Meaney E, Ceballos G, Gutiérrez-Salmeán G. Obesity, Metabolic Syndrome, and Dietary Therapeutical Approaches with a Special Focus on Nutraceuticals (Polyphenols): A Mini-Review. Int J Vitam Nutr Res. 2014;84(3-4):113-123. doi:10.1024/0300-9831/a000198
- Kwok KO, Fries LR, Silva-Zolezzi I, Thakkar SK, Iroz A, Blanchard C. Effects of Probiotic Intervention on Markers of Inflammation and Health Outcomes in Women of Reproductive Age and Their Children. Front Nutr. 2022;9:889040. doi:10.3389/fnut.2022.889040
- Gómez-Dantés O, Alonso-Concheiro A, Razo-García C, Lilia Bravo-Ruiz M, Orozco E, Serván-Mori E, Alpuche-Aranda C, Hernández-Ávila M, Híjar-Medina M, Lamadrid-Figueroa H, Medina-Mora ME, Mohar-Betancourt A, Reynales-Shigematsu LM, Rivera-Dommarco J, Riojas-Rodríguez H, Campillo-García JI, Lozano-Ascencio R, Martínez-Palomo A. Prioridades de Investigación En Salud En México. 1a. Cuernavaca, Morelos, México: Instituto Nacional de Salud Pública; 2017.
- Henn RE, Noureldein MH, Elzinga SE, Kim B, Savelieff MG, Feldman EL. Glial-neuron crosstalk in health and disease: A focus on metabolism, obesity, and cognitive impairment. Neurobiol Dis. 2022;170:105766. doi:10.1016/j.nbd.2022.105766
- Robb JL, Morrissey NA, Weightman Potter PG, Smithers HE, Beall C, Ellacott KLJ. Immunometabolic Changes in Glia – A Potential Role in the Pathophysiology of Obesity and Diabetes. Neuroscience. 2020;447:167-181. doi:https://doi.org/10.1016/j.neuroscience.2019.10.021
- Marelli C, Salsano E, Politi LS, Labauge P. Spinal cord involvement in adult-onset metabolic and genetic diseases. J Neurol Neurosurg Psychiatry. 2019;90(2):211 LP - 218. doi:10.1136/jnnp-2018-318666
- World Health Organization [WHO]. Obesity.
- World Health Organization [WHO]. ICD-11 for Mortality and Morbidity Statistics (Version: 02/2022). Online. https://icd.who.int/browse11/l-m/en#/http://id.who.int/icd/entity/1890900469. Published 2022. Accessed July 8, 2022.
- Lampe L, Zhang R, Beyer F, Huhn S, Kharabian Masouleh S, Preusser S, Bazin PL, Schroeter ML, Villringer A, Witte AV. Visceral obesity relates to deep white matter hyperintensities via inflammation. Ann Neurol. 2019;85(2):194-203. doi:10.1002/ana.25396
- Monteiro R, Sivasubramanian MK, Balasubramanian P, Subramanian M. Obesity-Induced Sympathoexcitation is Associated with Glial Senescence in the Brainstem. FASEB J. 2020;34(S1):1. doi:https://doi.org/10.1096/fasebj.2020.34.s1.06223
- Balasubramanian P, Branen L, Sivasubramanian MK, Monteiro R, Subramanian M. Aging is associated with glial senescence in the brainstem - implications for age-related sympathetic overactivity. Aging (Albany NY). 2021;13(10):13460-13473. doi:10.18632/aging.203111
- Salas-Venegas V, Flores-Torres RP, Rodríguez-Cortés YM, Rodríguez-Retana D, Ramírez-Carreto RJ, Concepción-Carrillo LE, Pérez-Flores LJ, Alarcón-Aguilar A, López-Díazguerrero NE, Gómez-González B, Chavarría A, Konigsberg M. The Obese Brain: Mechanisms of Systemic and Local Inflammation, and Interventions to Reverse the Cognitive Deficit. Front Integr Neurosci. 2022;16. doi:10.3389/fnint.2022.798995
- Gómez-Apo E, Mondragón-Maya A, Ferrari-Díaz M, Silva-Pereyra J. Structural Brain Changes Associated with Overweight and Obesity. J Obes. 2021;2021:6613385. doi:10.1155/2021/6613385
- Milte CM, Ball K, Crawford D, McNaughton SA. Diet quality and cognitive function in mid-aged and older men and women. BMC Geriatr. 2019;19(1):361. doi:10.1186/s12877-019-1326-5
- Douglass JD, Dorfman MD, Thaler JP. Glia: silent partners in energy homeostasis and obesity pathogenesis. Diabetologia. 2017;60(2):226-236. doi:10.1007/s00125-016-4181-3
- Zulfiqar S, Garg P, Nieweg K. Contribution of astrocytes to metabolic dysfunction in the Alzheimer’s disease brain. Biol Chem. 2019;400(9):1113-1127. doi:10.1515/hsz-2019-0140
- Hao S, Dey A, Yu X, Stranahan AM. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun. 2016;51:230-239. doi:10.1016/j.bbi.2015.08.023
- Cope EC, LaMarca EA, Monari PK, Olson LB, Martinez S, Zych AD, Katchur NJ, Gould E. Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J Neurosci. 2018;38(41):8889-8904. doi:10.1523/JNEUROSCI.0789-18.2018
- Sobesky JL, Barrientos RM, De May HS, Thompson BM, Weber MD, Watkins LR, Maier SF.. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain Behav Immun. 2014;42:22-32. doi:10.1016/j.bbi.2014.06.017
- Fujita Y, Yamashita T. The Effects of Leptin on Glial Cells in Neurological Diseases. Front Neurosci. 2019;13:828. doi:10.3389/fnins.2019.00828
- Rahman MH, Kim M-S, Lee I-K, Yu R, Suk K. Interglial Crosstalk in Obesity-Induced Hypothalamic Inflammation. Front Neurosci. 2018;12:939. doi:10.3389/fnins.2018.00939
- Zhao F-Q, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007;8(2):113-128. doi:10.2174/138920207780368187
- Wilson-O’Brien AL, Patron N, Rogers S. Evolutionary ancestry and novel functions of the mammalian glucose transporter (GLUT) family. BMC Evol Biol. 2010;10:152. doi:10.1186/1471-2148-10-152
- Yu S, Ding WG. The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res. 1998;797(1):65-72. doi:10.1016/s0006-8993(98)00372-2
- Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia. 1997;21(1):2-21. doi:10.1002/(SICI)1098-1136(199709)21:1<2::AID-GLIA2>3.0.CO;2-C
- Horikoshi Y, Sasaki A, Taguchi N, Maeda M, Tsukagoshi H, Sato K, Yamaguchi H. Human GLUT5 immunolabeling is useful for evaluating microglial status in neuropathological study using paraffin sections. Acta Neuropathol. 2003;105(2):157-162. doi:10.1007/s00401-002-0627-4
- Mizuno TM, Lew PS, Jhanji G. Regulation of the Fructose Transporter Gene Slc2a5 Expression by Glucose in Cultured Microglial Cells. Int J Mol Sci. 2021;22(23). doi:10.3390/ijms222312668
- Jurcovicova J. Glucose transport in brain - effect of inflammation. Endocr Regul. 2014;48(1):35-48. doi:10.4149/endo_2014_01_35
- Hendrix RD, Ou Y, Davis JE, Odle AK, Groves TR, Allen AR, Childs G V, Barger SW. Alzheimer amyloid-β- peptide disrupts membrane localization of glucose transporter 1 in astrocytes: implications for glucose levels in brain and blood. Neurobiol Aging. 2021;97:73-88. doi:10.1016/j.neurobiolaging.2020.10.001
- Fuente-Martín E, García-Cáceres C, Granado M, de Ceballos ML, Sánchez-Garrido MÁ, Sarman B, Liu Z-W, Dietrich MO, Tena-Sempere M, Argente-Arizón P, Díaz F, Argente J, Horvath TL, Chowen JA. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J Clin Invest. 2012;122(11):3900-3913. doi:10.1172/JCI64102
- Tomassoni D, Martinelli I, Moruzzi M, Micioni Di Bonaventura MV, Cifani C, Amenta F, Tayebati SK. Obesity and Age-Related Changes in the Brain of the Zucker Lepr (fa/fa) Rats. Nutrients. 2020;12(5). doi:10.3390/nu12051356
- Naranjo V, Contreras A, Merino B, Plaza A, Lorenzo MP, García-Cáceres C, García A, Chowen JA, Ruiz-Gayo M, Del Olmo N, Cano V. Specific Deletion of the Astrocyte Leptin Receptor Induces Changes in Hippocampus Glutamate Metabolism, Synaptic Transmission and Plasticity. Neuroscience. 2020;447:182-190. doi:https://doi.org/10.1016/j.neuroscience.2019.10.005
- Fuente-Martín E, García-Cáceres C, Argente-Arizón P, Díaz F, Granado M, Freire-Regatillo A, Castro-González D, Ceballos ML, Frago LM, Dickson SL, Argente J, Chowen JA. Ghrelin Regulates Glucose and Glutamate Transporters in Hypothalamic Astrocytes. Sci Rep. 2016;6(1):23673. doi:10.1038/srep23673
- Schüler R, Seebeck N, Osterhoff MA, Witte V, Flöel A, Busjahn A, Jais A, Brüning JC, Frahnow T, Kabisch S, Pivovarova O, Hornemann S, Kruse M, Pfeiffer AFH. VEGF and GLUT1 are highly heritable, inversely correlated and affected by dietary fat intake: Consequences for cognitive function in humans. Mol Metab. 2018;11:129-136. doi:10.1016/j.molmet.2018.02.004
- Hernandez-Garzón E, Fernandez AM, Perez-Alvarez A, Genis L, Bascuñana P, Fernandez de la Rosa R, Delgado M, Angel Pozo M, Moreno E, McCormick PJ, Santi A, Trueba-Saiz A, Garcia-Caceres C, Tschöp MH, Araque A, Martin ED, Torres Aleman I. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia. 2016;64(11):1962-1971. doi:10.1002/glia.23035
- Sasaki T. Neural and Molecular Mechanisms Involved in Controlling the Quality of Feeding Behavior: Diet Selection and Feeding Patterns. Nutrients. 2017;9(10). doi:10.3390/nu9101151
- Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, Shulman RG, Tamborlane WV. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci USA. 1992;89(3):1109-1112. doi:10.1073/pnas.89.3.1109
- Pardridge WM, Triguero D, Farrell CR. Downregulation of blood-brain barrier glucose transporter in experimental diabetes. Diabetes. 1990. doi:10.2337/diab.39.9.1040
- Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging. 1997;18(5):469-474. doi:10.1016/s0197-4580(97)00111-5
| Recibido: 12 de julio, 2022 | Aceptado: 02 de septiembre, 2022 |
*Correspondencia: Méndez-Flores OG. Universidad Juárez Autónoma de Tabasco, División Académica de Ciencias de la Salud, Villahermosa, Tabasco, México. CP. 86690. Tel. (993) 3581500 Ext. 6300. E-mail: orquidia.mendez@ujat.mx
Este es un artículo de libre acceso distribuido bajo los términos de la licencia de Creative Commons, (http://creamasal@unam.mxtivecommons.org/licenses/by-nc/3.0), que permite el uso no comercial, distribución y reproducción en algún medio, siempre que la obra original sea debidamente citada.
